











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
Los errores innatos del metabolismo son enfermedades genéticas que a nivel individual son raras, pero miradas globalmente afectan a cerca de 1:1.000 individuos. Se han descrito unas 600 entidades 1, aunque una reciente clasificación identifica 1,015, con 111 condiciones adicionales que no llegan a cumplir todos los criterios de inclusión 2. Un 43 % afecta al sistema nervioso central y un 70 % más de un sistema anatómico y funcional. Pueden debutar a cualquier edad y su variabilidad genética hace que tengan diferentes expresiones clínicas y bioquímicas 1.
La homocistinuria hace parte de este tipo de enfermedades y su importancia radica en que constituye un factor de riesgo para desordenes cardiovasculares y neurológicos, con arterioesclerosis prematura y trombosis, además de otras alteraciones 3.
Existen varias causas que elevan los niveles sanguíneos de homocisteina, como consecuencia de patologías genéticas y adquiridas. Dentro de estas últimas están la insuficiencia renal crónica, el hipotiroidismo, la edad avanzada, la toxicidad medicamentosa (fibratos, ácido nicotínico e inhibidores de la bomba de protones, metrotexate, óxido nitroso, antagonistas de los folatos), las deficiencias en la ingesta de vitamina B6, B12, el ácido fólico (cofactores en el metabolismo de la metionina y la homocisteina) 4,5) y el uso del cigarrillo 6.
Respecto a las causas genéticas, están los errores innatos del metabolismo, como deficiencia de homocisteina B-sintasa (CBS) (homocisteina muy elevada, metionina elevada, cistationina disminuida), la deficiencia de metionina S-adenosil transferasa hepática (leve aumento de la homocisteina, metionina elevada, S-adenosil metionina disminuida), deficiencia de glicina N-metiltransferasa (marcado aumento de metionina y adenosilmetionina, con homocisteina y cistationina normales o ligeramente elevadas), deficiencia de S-adenosil-homocisteina hidroxilasa (adenosilhomocisteina muy elevada, homocisteina levemente aumentada, metionina muy alta, cistationina levemente elevada), deficiencia de adenosin quinasa (metionina muy alta con S-adenosil homocisteina normal o levemente elevada), aciduria metilmalonica (metionina elevada, homocisteina levemente elevada, metilmalónico en orina elevado) 7,8. Niveles normales o bajos de metionina y elevación de la cistatonina en plasma y del ácido metilmalónico en orina sugieren que la causa de homocistinuria se debe a enfermedades distintas de la deficiencia de CBS 5.
Descripción del caso
Paciente de 13 años que consulta por cuadro de cuatro días de evolución de cefalea en hemicráneo izquierdo tipo peso asociada a fotofobia, náuseas y vomito. Ocho horas antes de la consulta aumentó la cefalea, y estuvo acompañada de disartria, afasia motora, hemiparesia izquierda y relajación del esfínter urinario. Dentro de los antecedentes de importancia es fruto de una primera gestación, embarazo controlado de transcurso normal, parto vaginal a las 40 semanas, peso al nacer de 2950 gr y talla de 51 cm; no requirió hospitalización. A los cinco años presentó apendicitis con peritonitis. A los nueve años oftalmología diagnosticó subluxación bilateral del cristalino y genética inicia estudio de síndrome de Marfan. Desde los once años presentaba episodios de cefalea manejados como migraña. Cuatro meses antes del episodio actual presentó un evento de cefalea intensa que se acompañó de hemiparesia izquierda y afasia, de la cual se recupera completamente en 24 horas. Se realizó una tomografía simple de cráneo (TAC) que fue interpretada como normal, valorada por neurología pediátrica, que diagnosticó migraña y formuló para esto naproxeno y ácido valproico. Su desarrollo psicomotor en la infancia temprana fue normal, pero en el momento de la consulta cursaba 6° grado con trastorno en el aprendizaje (había perdido en dos ocasiones el año). Los padres la describen de mal genio. Dentro de los antecedentes familiares, están los padres no consanguíneos, dos hermanos sanos, pero una hermana con talla alta, al igual que abuelos y tíos. No presenta antecedentes familiares de Marfan ni subluxación del cristalino.
Al ingreso manifestaba intenso dolor cervical, afasia motora, desviación de la mirada a la derecha, sialorrea, hemiparesia izquierda, Babinsky izquierdo, reflejo rotuliano izquierdo exaltado y no mostraba alteración en la sensibilidad ni signos meníngeos. Su talla es de 173 cm (por encima del percentil 95), delgada, extremidades largas y con aracnodactilia. El TAC cerebral simple evidenció trombosis de varios senos venosos (figura 1); el estudio se complementó con Angio TAC cerebral (figura 2), resonancia magnética cerebral (figura 3) y Angio resonancia cerebral contrastada (figura 4), que confirman la trombosis de los senos venosos. Después de diagnosticar accidente isquémico se trasladó a la unidad de cuidados intensivo (UCI) y se anticoagula con enoxaparina. No necesitó soporte inotrópico ni ventilatorio y posteriormente se llevó a pisos. El ecocardiograma y el doppler carotideo fueron normales. No se encontró dislipidemia y el estudio de enfermedad autoinmune fue negativo. Los exámenes para trombofilia, tomados en etapa aguda, solo mostraron una proteína S ligeramente disminuida (57 %, VN de 59 a 118 %), al igual que la antitrombina m (58,8 %, VN 83-128 %). Se ampliaron estudios con ácido fólico y vitamina B12 en sangre, al igual que los ácidos orgánicos en orina por cromatografía; todos estaban normales. Los aminoácidos en sangre mostraron elevación de la metionina (63 µmol/L, VN de 10 a 45 µmol/L) y la homocisteina (3 µmol/L, VN de 0 a 1 µmol/L) con cistationina baja (0, VN de 0 a 3 µmol/L). El estudio de homocisteina en sangre fue de 214 µmol/L (VN en mujer de 5,08 A 15,39 µmol/L), lo que confirmó el diagnóstico de homocistinuria por deficiencia de CBS.
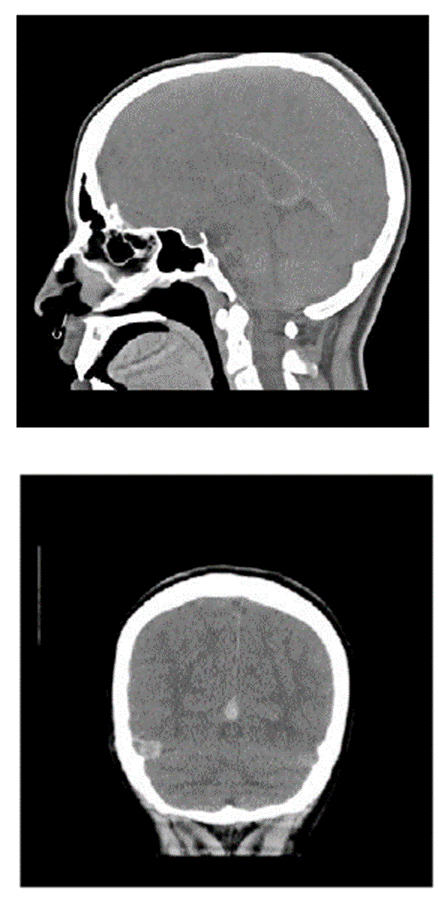
Fuente: Hospital El Tunal. Nota: muestra trombosis venosa aguda de vena de galeno, cisterna, vena supracallosa y seno transverso del lado derecho, trayecto vascular venoso.
Figura 1 Tomografía axial computarizada (TAC) de cráneo simple.
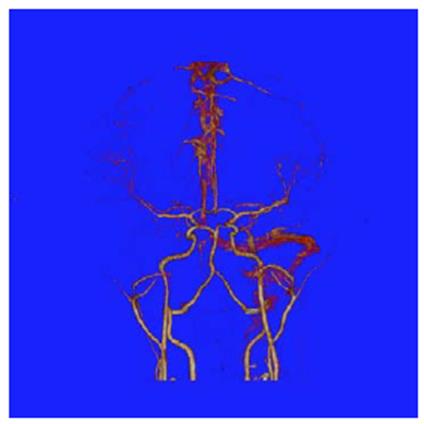
Fuente: Hospital El Tunal. Nota: reporta defectos de llenado con el contraste comprometiendo el curso parietal y occipital del seno longitudinal superior, del seno sagital inferior, la vena cerebral interna, la vena de galeno, seno recto, tórcula de Herófilo, seno transverso derecho, seno sigmoideo derecho y aparentemente seno petroso inferior derecho.
Figura 2 AngioTAC cerebral.
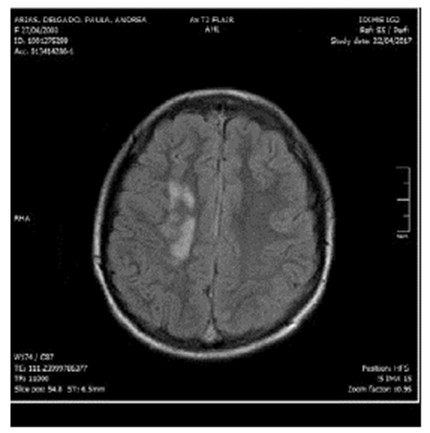
Fuente: IDIME. Nota: evidencia signos de trombosis de senos venosos con compromiso del seno longitudinal superior y transverso derecho, lesión parenquimatosa del centro semioval derecho, relacionable con infarto venoso.
Figura 3 Resonancia magnética cerebral
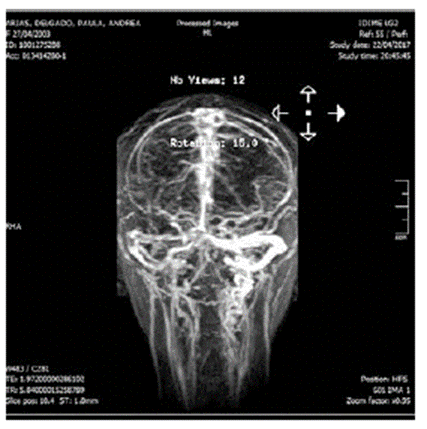
Fuente: IDIME. Nota: muestra signos de trombosis venosa comprometiendo el seno longitudinal superior y parcialmente el seno transverso derecho.
Figura 4 Angioresonancia cerebral contrastada
Se inició tratamiento con piridoxina 150 mg cada 8 horas y ácido fólico 5 mg una vez al día. Al egreso la recuperación neurológica fue completa. En el seguimiento ambulatorio la paciente continua asintomática. El control de la homocisteina, casi dos meses después de iniciado el manejo, fue de 124 µmol/L, lo que la hace una respondedora parcial a la piridoxina; por esta razón, se decide aumentar la dosis a 200 mg cada 8 horas, una dieta baja en metionina y rica en cisteina. Está pendiente un nuevo control de homocisteina para definir si es necesario adicionar betaina, e igualmente la realización del estudio genético directo de las mutaciones más frecuentes descritas en Colombia.
Discusión
La homocisteina no forma parte de las proteínas alimentarias, siendo sintetizada a partir de la metionina. En la homocistinuria, debido a la deficiencia de la cistationina B-sintasa (OMIN 236200), no se lleva a cabo la reacción por la cual la homocisteina en conjunto con la serina produce cistationina, lo que ocasiona elevación de la homocisteina y metionina, con disminución de la cistina y cistationina. Constituye el error congénito más frecuente de los aminoácidos sulfurados y su gen está localizado en el cromosoma 21 (21q22.3), 9,10 (figura 5).
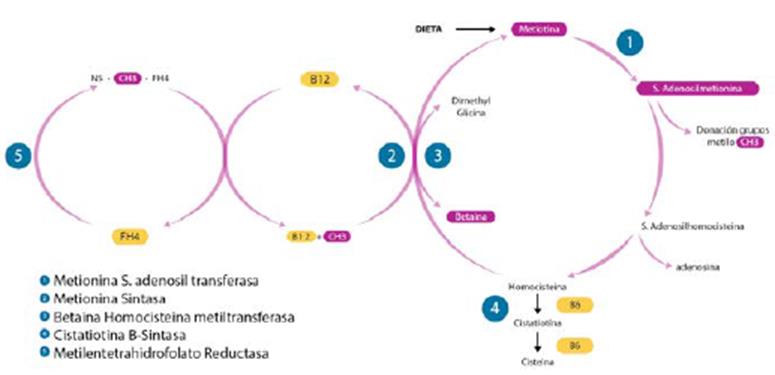
Fuente: elaboración propia a partir de (6 y 9).
Figura 5 Deficiencia de cistationina-b sintasa. Vías metabólicas
La incidencia varía según la región, por ejemplo, en Irlanda y Australia es de 1/60.000; en España, de 1/200.000-300.000 8, y el Japón, de 1/90.000 9; pero en Qatar puede llegar a ser de 1:1.800 debido a altas tasas de consanguinidad, con un 2 % de portadores 5. Los estudios de cribado neonatal hacen pensar que es mucho más frecuente de lo que se pensaba, y podría presentarse en 1/20.000 recién nacidos 11. Estudios moleculares epidemiológicos predicen frecuencias entre 1:6.400 y 1:20.500 en poblaciones de Europa 12. La frecuencia de heterocigotos es menor que 1-2 % de la población general 8.
La herencia es autosómica recesiva 13, y existe gran variabilidad bioquímica dada por una amplia diversidad de mutaciones 14, lo que explica las diferentes edades de presentación y grados de severidad 5. Han sido reportadas más de 180 mutaciones, y la mayoría de ellas han sido puntuales (72 %) 10; además, existe mayor correlación entre el genotipo y el fenotipo cuando las mutaciones son homocigotas que heterocigotas compuestas 5. En los respondedores (por la presencia de actividad enzimática residual) como en los no respondedores a la piridoxina, las mutaciones pueden ser homocigotas o heterocigotas compuestas. Dentro de las que no responden están la p.G307S y la p.T191M, y de las respondedoras, la p.I278T 8. En España, Portugal y Colombia es frecuente la mutación p.T191M. La prevalencia de p.T191M entre los alelos mutantes en CBS en los diferentes países fue: 0,75 en Colombia, 0,52 en España, 0,33 en Portugal, 0,25 en Venezuela, 0.20 en Argentina y 0.14 en Brasil. No se encontró correlación entre el genotipo y el fenotipo 15.
Los recién nacidos son normales, pero posteriormente tendrán alteraciones en el crecimiento y retraso en el desarrollo 16. La enfermedad es de inicio lento y progresivo, y puede afectar muchos sistemas 8. En los respondedores a la piridoxina, sus manifestaciones generalmente serán más leves y de presentación tardía 12, aunque también se han observado diferentes fenotipos en hermanos 17. Una de las características fenotípicas de esta enfermedad son las alteraciones esqueléticas y sus rasgos marfanoides, presentes en la paciente. La homocistinuria hace parte de las tallas altas, conformadas por un grupo heterogéneo de entidades que van desde la normalidad, endocrinopatías, cromosomopatías, síndromes mendelianos o metabolopatías. Su aproximación diagnóstica se basará en una buena anamnesis y un completo examen físico, que orientarán la solicitud de estudios complementarios 18. Las alteraciones del tejido conectivo en la homocistinuria se deben a la inhibición de la síntesis del colágeno y de la elastina, lo que genera alteraciones en la piel, las articulaciones y el esqueleto 8. La osteoporosis puede presentarse desde el año de edad 7 -afecta a casi todos los pacientes-, y generar escoliosis y fracturas patológicas. Los individuos afectados son altos, principalmente por unas extremidades largas (dolicoestenomelia), con una relación del segmento superior/inferior disminuida 8. Los cambios cualitativos de la fibrilina es posible que generen una deficiencia en los factores necesarios para prevenir la elongación de los huesos, y esto ocasiona el hipercrecimiento 18. La aracnodactilia está presente en el 50 % de los pacientes. Otras alteraciones descritas son genu valgo, pectum excavatum o carinatum, pies cavos, limitación de la movilidad articular -especialmente en las extremidades-, osteoporosis con vértebras biconcavas, espículas metafisiarias en manos y pies, elongación de los huesos del carpo, 4 metacarpiano corto y retraso del desarrollo del semilunar 8. En la homocistinuria la edad ósea corresponderá a la edad cronológica 19. Los pacientes respondedores a la piridoxina, comparados con los no respondedores, tendrán un menor porcentaje de anomalías esqueléticas 20.
La luxación del cristalino es otra característica de la homocistinuria, la cual se diagnosticó a los nueve años en la paciente que describimos. Esta luxación puede tener causas congénitas (ectopia lentis) o adquiridas, mientras que en la homocistinuria es bilateral e inferonasal; esto la diferencia del síndrome de Marfan, siendo acá superior y bilateral. Otras entidades en las cuales está presente son el síndrome de Weill-Marchesani (microesferofaquia) 21, el síndrome de Ehlers Danlos 5, la deficiencia de sulfito oxidasa y la deficiencia del cofactor del molibdeno 22. La luxación del cristalino en la homocistinuria se debe a alteraciones en la estructura de la fibrilina, y se presenta en etapas tempranas de la vida; afecta más o menos al 80 % de las personas a los diez años 8. Su presencia y el retardo en el desarrollo hacen sospechar la posibilidad de tener la enfermedad 5. El temblor del iris (iridodonesis) se debe a la falta de su soporte habitual y con frecuencia orienta al diagnóstico de la enfermedad. Otras alteraciones oculares descritas en esta enfermedad son la miopía (que es rápidamente progresiva), el glaucoma, el desprendimiento y la degeneración de la retina, las cataratas y la atrofia óptica 8.
El motivo de consulta de la paciente fue la cefalea asociada a alteraciones neurológicas, que fueron ocasionadas por su accidente cerebrovascular. Los accidentes cerebrovasculares, en general, vienen siendo descritos con mayor frecuencia en la población infantil, posiblemente por mejores técnicas de imagen, así como por el reconocimiento de un número mayor de causas 23. En ocasiones se diagnostican de manera fortuita al realizarse una imagen cerebral por otro motivo 24. Cuando se presentan deben hacerse los estudios de trombofilia correspondientes (dentro de ellos la homocisteina) e iniciar el manejo adecuado 24. Pueden tener una incidencia acumulada de secuelas neurológicas entre el 8 % y el 47 %, con una mortalidad en el 17 % de los casos 24. En la homocistinuria la trombosis es secundaria al daño vascular ocasionado por la elevación plasmática de la homocisteina, que genera toxicidad sobre el endotelio vascular, mayor adherencia plaquetaria, proliferación de las células musculares lisas 8, activación de la cascada de la coagulación, incremento de la oxidación de partículas LDL y depósito de estas en la pared arterial 4.
Un estudio realizado en Colombia mostró que los afectados por homocistinuria presentaron un aumento significativo del inhibidor del activador del plasminógeno tipo 1 25. El compromiso venoso es más frecuente que el arterial y puede afectar cualquier parte del cuerpo, lo cual se constituye en la principal causa de morbilidad y mortalidad temprana 5. El riesgo estimado de tromboembolismo venoso para la hiperhomocisteinemia es 2,5 veces mayor comparado con la población general, de 3 para la mutación de la protrombina 20210 heterocigota y de 80 para la mutación del factor V Leiden homocigota 24. La coexistencia de homocistinuria con el déficit del factor V de Leiden aumenta más el riesgo de trombosis 26.
Hay una arterioesclerosis prematura y el 50 % de los pacientes no tratados presenta complicaciones vasculares antes de los 30 años. Las complicaciones tromboembólicas afectan arterias y venas, ya sean grandes o pequeñas, en cualquier localización, independientes de la edad. El compromiso de las arterias carótidas y renales son causa frecuente de mortalidad, pero la tromboflebitis y el embolismo pulmonar son los accidentes vasculares más frecuentes 8.
En un estudio la trombosis venosa profunda representó el 51 % de los casos; los accidentes cerebrovasculares, el 32 %; el tromboembolismo pulmonar, el 25 %; la enfermedad vascular periférica, el 11 % y el infarto agudo de miocardio, el 4 % 27. Es importante aclarar que no todos los pacientes que con homocisteinemia y eventos trombóticos presentan los hallazgos clásicos de la homocistinuria 28, como las manifestaciones óseas, oculares o neurológicas 29. Los respondedores a la piridoxina, comparados con los no respondedores tendrán un menor porcentaje de eventos trombóticos 20. El manejo de la trombosis venosa cerebral se hace con heparina o heparina de bajo peso molecular durante tres meses, así el seno este re-canalizado, pero si este no es el caso se prolonga a seis meses 30. Dependiendo de la gravedad de la trombosis, algunos médicos han asociado a la heparina intravenosa el factor recombinante activador del plasminógeno tisular, administrado intravenosamente durante 36 horas hasta la restauración del flujo venoso, con buenos resultados y sin complicaciones significativas 3. Nuestro caso se manejó solo con heparina de bajo peso molecular, con una muy buena respuesta clínica, pues se presentó a los pocos días una recuperación completa de las funciones neurológicas perdidas.
En sus primeros años la paciente tuvo un desarrollo psicomotor normal, pero luego empezó a presentar trastorno del aprendizaje con pérdidas escolares, y fue descrita de mal genio. Cerca del 60 % de los pacientes presenta retraso mental, generalmente no grave y con afectación variable. Este se debe a la inhibición competitiva del transporte de los aminoácidos al cerebro y la formación de neurotransmisores por la elevada concentración de homocisteina y metionina 8 y por el déficit de cistationina, importante en el cerebro 31. Los coeficientes de inteligencia varían entre 10 y 138 31, con un promedio de 80, siendo menor en los pacientes no respondedores a la piridoxina 32. En los niños que inician un tratamiento desde el nacimiento y mantienen un adecuado control de los niveles de homocisteina el coeficiente intelectual será normal, contrario a aquellos que tienen un inadecuado control o el tratamiento se inicia tardíamente, en estos será menor 33. Puede haber trastornos de la personalidad esquizoide y actividad electroencefalográfica con ondas muy lentas 8. Los trastornos psiquiátricos y del comportamiento se llegan a presentar en más del S0 % de los casos, y las convulsiones pueden estar en el 20 % de los pacientes 16. Los signos extrapiramidales son raros 31.
En la homocistinuria hay una elevación de homocisteina y metionina en el plasma, con disminución de cistina y cistationina 8, concordante con los hallazgos de la paciente. Se debe medir la homocisteina total, que incluye la homocisteina unida a proteínas (70-80 y), homocisteina disulfuro y homocisteina libre (que se oxida a homocistina) 8, pues los niveles de homocisteina libre tienen una menor sensibilidad y reproducibilidad (S). Por lo general, los valores de homocisteina total serán mayores de 100 µmol/L (en la paciente estaban en 214 µmol/L), aunque valores menores no descartan la enfermedad, ya que se pueden dar en algunas variantes.
Para lo anterior es importante tener presente si se están tomando suplementos de piridoxina, pues los pacientes respondedores a esta vitamina pueden llegar a normalizar sus valores. Cuando esta es la situación se debe suspender la piridoxina por lo menos dos semanas antes e idealmente entre uno y dos meses (S). Los heterocigotos tendrán niveles de homocisteina ligeramente elevados, en general entre 1S-30 µmol/L (valor normal entre S y 1S µmol/L) 7. La medición de la homocisteina en orina es menos sensible que la sérica (S). La hiperhomocisteinemia se ha clasificado en moderada (1S a 30 µmol/L), intermedia (entre 30 y 100 µmol/L) y severa (mayor de 100 µmol/L 34; esta última es el caso de la paciente que describimos. Se deben estudiar los ácidos orgánicos en orina para ver si hay aciduria metilmalónica, presente en la homocistinuria de algunos defectos del metabolismo de la cobalamina, donde se encontrará aumento de ácido metilmalónico en la orina (en la paciente fue normal este examen). Para la confirmación de la enfermedad se valora la actividad enzimática de la CBS en cultivo de fibroblastos, en tejido hepático o leucocitos; también se puede llevar a cabo un estudio genético directo, aunque si se desconoce el tipo de mutación se puede hacer un estudio genético indirecto 8. Ninguna técnica demuestra todas las anormalidades en todos los casos 5. El diagnóstico prenatal en el primer trimestre se puede hacer mediante técnicas moleculares si se conocen las mutaciones en los padres; si no, la actividad enzimática se determinaría en cultivo de amniocitos, no en vellosidades coriónicas 5,8. El screening neonatal se basa en la elevación de la metionina. Se ha sugerido un punto de corte entre 39 a 50 µmol/L para aumentar su sensibilidad diagnóstica, aunque los pacientes respondedores a piridoxina posiblemente presenten falsos negativos, por lo que se aconseja hacer la relación metionina/fenilalanina. Como la metionina se puede elevar en otras enfermedades, para aumentar la especificidad se mide la homocisteina total como segundo marcador 5. Este tamizaje en recién nacidos no se hace midiendo la homocisteina, ya que es técnicamente más difícil y su costo mayor. También se hace el estudio neonatal mediante genética molecular en poblaciones de alto riesgo y con un limitado número de mutaciones prevalentes 12. Cuando un miembro de la familia presenta la enfermedad, se debe ofrecer el estudio a los demás integrantes 5.
La paciente ha sido manejada con piridoxina, ácido fólico y recomendaciones dietéticas, además de la anticoagulación con enoxaparina. En el tratamiento se busca normalizar los niveles de homocisteina; su inicio temprano disminuye la morbilidad, los eventos trombóticos, la subluxación del cristalino y mejora el coeficiente intelectual 12, en especial si se hace en el periodo neonatal. En la dieta se disminuye la ingesta de metionina, pero solo si con la piridoxina no se logra reducir los niveles de homocisteina 5. Como la producción endógena de cistina está bloqueada, este aminoácido se convierte en esencial (es precursor de la taurina y el glutatión), por lo cual se requieren suplementos de L-cistina.
También se deben controlar los niveles de serina y suplementarlos si hay necesidad 8. Con estas dietas bajas en proteínas y como los aminoácidos son esenciales para la formación de proteínas, es necesario el aporte de suplementos nutricionales especiales que no contengan metionina. La piridoxina es precursora del cofactor del fosfato de piridoxal, siendo necesaria para la actividad de la CBS 8. Entre el 15 % y el 50 % de los pacientes responden total o parcialmente a esta vitamina (dependiendo de la variable genética y, por ende, de la actividad enzimática residual) 8,17, y normalizan o disminuyen la hiperhomocisteinemia y la hipermetioninemia. Si no responden, se aconseja, sin embargo, continuar la administración de la vitamina, pues a largo plazo disminuyen los episodios de tromboembolismo.
La dosis por administrar es la que mantiene los niveles de homocisteina más bajos. La dosis que se recomienda inicialmente es de 10 mg/kg/día, con un máximo de 500 mg al día y un mínimo de 100 mg. Se consideran respondedores cuando los niveles de homocisteina total están por debajo de 50 µmol/L, con lo cual no se necesita otro tratamiento; respondedores parciales cuando la homocisteina disminuye más del 20 %, pero su nivel está por encima de 50 µmol/L, y por esto se necesita adicionar dieta o betaina, y no respondedores cuando disminuyen menos del 20 % 5.
Un paciente no debe ser considerado no respondedor a la piridoxina hasta que haya recibido de 500 a 1000 mg al día por al menos seis semanas 8. Algunos médicos indican una dosis en lactantes de 100 a 250 mg/día, en escolares entre 200 y 500 mg/día y para adolescentes y adultos de 500 a 1200 mg/día 9. Con dosis generalmente más altas de 1000 mg/día puede presentarse neuropatía sensorial. Junto a la piridoxina se debe dar ácido fólico entre 5 a 10 mg/día o ácido folínico entre 1 a 5 mg/día, ya que este favorece la remetilación de la homocisteina a metionina, así como la vitamina B12, inicialmente 1 mg intramuscular; es importante vigilar su posible deficiencia, y suplementarla si esto sucede. Cuando las medidas anteriores no han funcionado se administra betaina (derivada de la colina) como donante de grupos metilo, que activa la betaina: homocisteina metiltransferasa, remetilando la homocisteina y formando metionina. Cuando los niveles de metionina aumentan mucho pueden causar edema cerebral, especialmente si son mayores de 1000 µmol/L, por lo que deben vigilarse 10. Se recomienda iniciar la betaina a 100 mg/kg/día dividida en 2 dosis y aumentar semanalmente en 50 mg/kg, hasta un máximo de 150 a 200 mg/kg/día. La dosis usual del adulto es de 3 gramos cada 12 horas 5, aunque otros consideran 6-9 gramos al día en 2 o 3 dosis 10 (figura 6).
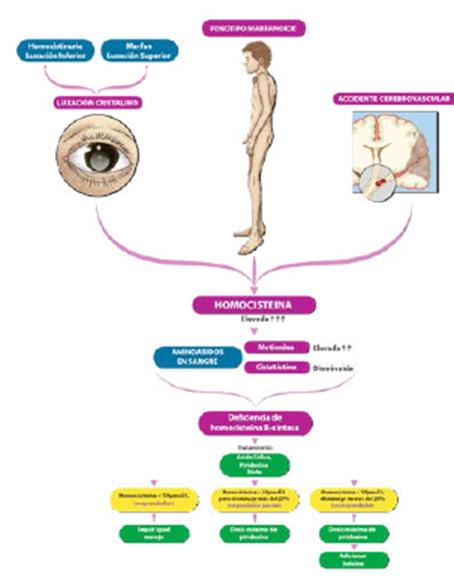
Se considera que un adecuado control en los respondedores a piridoxina equivale a un nivel sérico de homocisteina total por debajo de 50 µmol/L y en los no respondedores de 120 µmol/L (idealmente por debajo de 100 µmol/L). En los parcialmente respondedores, para llegar a tener niveles por debajo de 50 µmol/L se necesita adicionar una dieta baja en metionina, pero cuando los niveles séricos de este aminoácido llegan a estar por debajo de lo normal es posible que se afecten el crecimiento y neurodesarrollo del niño 5. La administración de vitamina C se utiliza para disminuir el riesgo de tromboembolismo a largo plazo, al mejorar la disfunción endotelial 35. Como medidas preventivas del tromboembolismo se debe evitar la deshidratación, el uso de anticonceptivos y las cirugías 8.
En el seguimiento se monitoriza la homocisteina durante uno o dos meses después de iniciar la piridoxina, y si es de los no respondedores, nuevamente de uno a dos meses después de iniciar la betaina. Hay que hacer seguimiento del estado nutricional, los niveles plasmáticos de aminoácidos (junto con la metionina), la vitamina B12 y el ácido fólico. En pacientes respondedores a piridoxina los controles posteriores pueden ser cada seis meses, pero en los no respondedores posiblemente se necesiten con más frecuencia 5. Son importantes los seguimientos nutricionales, oftalmológicos, neurológicos, densitometría ósea, cardiovasculares, psicológicos 8.
Sin tratamiento el pronóstico de la enfermedad es malo, y un 25 % de los pacientes fallece antes de los 30 años por vasculopatías. Con el tratamiento, especialmente en los respondedores a la piridoxina, las manifestaciones clínicas serán más leves y en los no respondedores, el manejo con betaína hará una evolución clínica aceptable 8.
Conclusiones
La homocistinuria no es una enfermedad frecuente y en Colombia se desconoce su prevalencia. El Ministerio de Salud y Protección Social la considera una enfermedad huérfana. Sus manifestaciones no siempre son típicas y posiblemente en muchas ocasiones pasan desapercibidas; en otras se llega al diagnóstico cuando ya se han presentado algunos episodios que pueden poner en peligro la vida de los pacientes, como el caso que describimos en este artículo. Lo ideal es hacer tamizaje universal, como se hace en otros países, ya que dicha enfermedad tiene un manejo que puede cambiar su pronóstico. Si esto no fuese posible, se deberían realizar estudios que orienten acerca de la prevalencia de la enfermedad e indicar aquellos grupos de mayor riesgo donde se pueda hacer una intervención temprana.

