











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
La atrofia muscular espinal (AME) se define como un conjunto de trastornos neurodegenerativos hereditarios, que afectan las células del asta anterior de la médula espinal y los núcleos motores en la parte inferior del tronco encefálico. Se caracteriza por debilidad y atrofia muscular simétrica progresiva, cuya gravedad depende de la edad de inicio y de complicaciones debilitantes a largo plazo que amenazan la vida de los pacientes 1-4.
En la Resolución 023 de 2023 (Ministerio de Salud y Protección Social, 2023) “por la cual se actualiza el listado de enfermedades huérfanas y raras en Colombia”, se encuentra la AME, como enfermedad huérfana, en relación con su baja prevalencia, cronicidad y alta complejidad. Se clasifica en AME asociada a variante genética en el brazo largo del cromosoma 5 (AME 5q) y AME no 5q 2.
La AME 5q representa del 80 al 90 % de los trastornos hereditarios de la motoneurona, con una incidencia de 1 en 6000-10000 nacidos vivos. Es causada por la deleción del gen de supervivencia de la motoneurona (SMN1) ubicado en el cromosoma 5q11.2-13.3, considerado el determinante de la enfermedad en razón a que su ausencia o variantes genéticas patogénicas, confirman la relación genotipo-fenotipo. El gen de supervivencia de la motoneurona 2 (SMN2), homólogo de SMN1, es modificador fenotípico 5-6.
La AME no 5q, por su parte, es de difícil precisión en cuanto a su frecuencia epidemiológica debido a la gran variabilidad de genes involucrados. En Colombia según el Instituto Nacional de Salud en el periodo epidemiológico 2016-2020 se notificaron 2159 casos de enfermedades del sistema nervioso, entre los cuales se identificaron tres de AME no 5q es decir, el 0,04 % de los 8555 registrados como huérfanas y raras 7. Durante 2021 las enfermedades del sistema nervioso notificadas por al Sistema de Vigilancia en Salud Pública (Sivigila) fue de 1187 casos, de las cuales 4949 fueron enfermedades huérfanas, sin especificar cuáles corresponden a cada caso 8. En el boletín epidemiológico de 2022 se notificaron al Sivigila 1368 casos de enfermedades del sistema nervioso que incluyeron nueve casos de otras AME no especificadas, lo que representa el 0,14 % de los 6657 casos de enfermedades huérfanas 30.
Los genes asociados con la AME no 5q se han expandido rápidamente debido al advenimiento de las tecnologías de secuenciación de nueva generación. Suelen clasificarse genéticamente según el patrón de herencia en: autosómico dominante, autosómico recesivo o ligado al cromosoma X y la distribución de la debilidad muscular en: proximal, distal o bulbar 9-11 (tablas 1 y 2).
Tabla 1 Características genotípicas y fenotípicas asociadas a la presentación de ame no 5q de inicio temprano
| Gen | OMIM | Nombre | Localización cromosómica | Función | Clínica e inicio de sintomatologia | Herencia | Condición | |
|---|---|---|---|---|---|---|---|---|
| Inicio temprano | ||||||||
| SMAX2 | UBA1 | 314370 | Modificador similar a la ubiquitina que activa la enzima 1 | Xp11.3 | Catalizar primer paso en conjugación de ubiquitina | Antenatal. Hipotonía, arreflexia, deformidades torácicas, rasgos dismórficos faciales, contracturas articulares, fracturas óseas, anomalías genitales | Recesivo ligado a cromosoma X | Atrofia muscular espinal infantil letal con artrogriposis, fracturas congênitas |
| BVVLS1 | SLC52A3 | 613350 | Familia de transportadores de solutos 52 miembro 3 | 20p1.3 | Codificar proteína transportadora de riboflavina | Infancia precoz-adultez. Debilidad de los brazos, manos y cara; ataxia; disfagia; atrofia lingual, parálisis pontobulbar, sordera neurosensorial | Autosomico recesivo | Síndrome de Brown Vialetto-Van Laere 1 |
| BVVLS2 | SCL52A2 | 613350 | Familia de transportadores de solutos 52 miembro 2 | 8q24.3 | Codificar proteína transportadora de riboflavina | Infancia precoz-adultez. Debilidad de brazos, manos y cara; ataxia; disfagia; atrofia lingual, parálisis pontobulbar, sordera neurosensorial | Autosomico recesivo | Síndrome de Brown Vialetto-Van Laere 2 |
| PCH1A | VRK1 | 602168 | Vaccinia relacionado con serina/treonina quinasa 1 | 14q32.2 | Codificar proteínas quinasasserina/ treonina con vaccinia | Infancia precoz. Microcefalia, hipotonía severa, arreflexia, discapacidad visual central, disfagia, insuficiencia respiratoria | Autosomico recesivo | Hipoplasia pontocerebelosa con atrofia muscular infantil |
| PCH1B | EXOSC3 | 606489 | Exosoma componente 3 | 9p13.2 | Codificar componente 3 no catalítico del exosoma humano | Infancia precoz. Microcefalia, retardo mental, muerte temprana | Autosomico recesivo | Hipoplasia pontocerebelosa con atrofia muscular infantil |
| PCH1C | EXOSC8 | 607596 | Exosoma componente 8 | 13q13.3 | Codificar componente 8 no catalítico del exosoma humano | Infancia precoz. Microcefalia, retardo mental, muerte temprana | Autosomico recesivo | Hipoplasia pontocerebelosa con atrofia muscular infantil |
| SMAPME | ASAH1 | 613468 | N-acilesfingosina amidohidrolasa 1 | 8p22 | Codificar familia de proteínas ceramidasas ácidas | Niñez temprana. Debilidad muscular proximal, hipotonía, arreflexia, atrofia muscular, fasciculaciones, pérdida auditiva neurosensorial, debilidad en músculos respiratorios, microcefalia | Autosomico recesivo | Atrofia muscular espinal con epilepsia mioclónica |
| LAAHD | GLE1 | 603371 | Mediador de exportación de arnGLEI | 9q34.11 | Codificar polipéptido de homólogo con levadura Glelp | Antenatal. Inmovilidad fetal, hidropesía, micrognatia, hipoplasia pulmonar, contracturas articulares | Autosomico recesivo | Artrogriposis letal con enfermedad de células del asta anterior |
| SMARD1 | IGHMBP2 | 604320 | Inmunoglobulina helicasa de unión a μ proteína 2 | 11q13 | Codificar miembro de superfamilia de helicasas | Infancia precoz. Debilidad muscular distal y en extremidad inferior, debilidad diafragmática precoz | Autosomico recesivo | Atrofia muscular espinal con parálisis de diafragma |
| SMARD2 | LAS1L | 309585 | Lanosterol sintasa 1 | Xq12 | Activar la actividad de unión al arn | Infancia precoz. Debilidad muscular distal y en extremidad inferior, debilidad diafragmática precoz | Recesivo ligado a cromosoma X | Atrofia muscular espinal con parálisis de diafragma |
| DSMA | TRPV4 | 600175 | Receptor transitorio potencial catiónico subfamilia V miembro 4 | 12q24.11 | Codificar familia de canal catiónico no selectivo permeable a Ca2 | Congénita. Debilidad muscular, contracturas distal y proximal de las piernas, hipoacusia neurosensorial | Autosomico dominante | Atrofia muscular espinal congênita de extremidades inferiores |
| SMALED1 | DYNC1H1 | 600112 | Dineína citoplasmática 1 cadena pesada 1 | 14q32.31 | Codificar familia de cadenas pesadas de dineína citoplasmática | Congénito hasta adultez. Deformidad muscular proximal en piernas respeta aductores y semitendinoso, no progresiva | Autosomico dominante | Atrofia muscular espinal en extremidades inferiores-1 |
| SMALED2 | BICD2 | 609797 | Adaptador de carga BICD 2 | 9q22.31 | Motilidad hacia extremo negativo por dineína en microtúbulos | Congénito hasta adultez. Progresión lenta, debilidad muscular proximal en piernas con algunas contracturas | Autosomico dominante | Atrofia muscular espinal de predominio en extremidades inferiores-2 |
Fuente: Peeters K. Clinical and genetic diversity of SMN1-negative proximal spinal muscular atrophies. Brain. Atrofias musculares espinales no asociadas a SMN1. Revista Médica Clínica Las Condes.
Tabla 2 Características genotípicasy fenotípicas asociadas a la presentación de ame no 5q de inicio tardío
| Tipo | Gen | OMIM | Nombre | Localización cromosómica | Función | Clínica | Herencia | Condición |
|---|---|---|---|---|---|---|---|---|
| Inicio tardío | ||||||||
| HMSNP | TFG | 602498 | Tráfico de retículo endoplasmático al regulador de Golgi | 3q12.2 | Codificar oncoproteínas de fusión | Adultos jóvenes. Parestesias musculares dolorosas, miotonía en manos, disfagia, debilidad muscular, atrofia muscular proximal o distal, fasciculaciones, posterior deterioro sensorial distal lentamente progresivo | Autosomico dominante | Neuropatía sensitivo-motora hereditaria proximal, tipo Okinawa |
| LGMD1B | LMNA | 150330 | Lamin A/C | 1q22 | Codificar proteína de la lámina nuclear | Adultos jóvenes. Debilidad muscular proximal progresiva y miocardiopatía | Autosomico dominante | Atrofia muscular espinal proximal de inicio en adultos seguida de afectación cardíaca |
| SMAX1 | AR | Sin OMIM | Receptor andrógeno | Xq12 | Codificar receptor androgénico | Adultez. Debilidad proximal, bulbar, trastorno endocrino | Recesivo ligado a cromosoma X | Enfermedad de Kennedy, atrofia bulbo-espinal ligada a X |
| SMAJ | CHCHD10 | 615903 | Dominio que contiene 10 Coiled- Coil-Helix-Coiled- Coil-Helix | 22q11.23 | Codificar proteínas mitocondriales | Adultez. Parestesias dolorosas, fasciculaciones, debilidad muscular yarreflexia | Autosomico dominante | Atrofia muscular espinal tipo jokela |
| ALS4 | SMAFK | 608465 | 606873 | 9q34.13 | Codificar proteína Sen1p | Inicio juvenil o adultez. Debilidad proximal y distal, temblor de manos, reflejos vivos | Autosómico dominante | Atrofia muscular espinal de inicio juvenil o adultez con características piramidales |
| SPSMA | TRPV4 | 605427 | Miembro 4 de la subfamilia V de canales catiónicos de potencial de receptor transitorio | 12q24.11 | Codificar un miembro de la subfamilia del canal de potencial receptor transitorio | Adultez temprana. Debilidad progresiva de los músculos faciales y pectorales con parálisis laríngea, sordera neurosensorial, anomalías esqueléticas que preservan gastronecmio medial y bíceps femoral | Autosómico dominante | Atrofia muscular espinal Escápulo-peroneal |
| SMAFK | VAPB | 605704 | Proteínas B y C asociadas a proteína similar a la sinaptobrevina (VAMP) | 20q13.32 | Codificar proteína de membrana de tipo IV | Adultez. Parestesias y fasciculaciones musculares | Autosómico dominante | Atrofia muscular espinal de inicio tardío tipo Finkel |
| SMAFK | HEXB | 606873 | Subunidad beta de hexosaminidasa | Subunidad beta de hexosaminidasa | Codifica subunidad beta de hexosaminidasa | Inicio adulto tardío. Debilidad muscular proximal de extremidades inferiores | Autosómico recesivo | Atrofia muscular espinal pura de inicio adulto tardío |
Fuente: Peeters K, 2014.
Los mecanismos fisiopatológicos secundarios al daño de la motoneurona incluyen: anomalías en la reparación del ADN (ácido desoxirribonucleico) (UBA1), alteración en procesamiento y degradación del ARN (ácido ribonucleico) (EXOSC3, EXOSC8), captación de vitaminas (SLC52A3 y SL- C52A2), regulación del ciclo celular (VRK1, TFG), metabolismo lipídico (ASAH1), transporte nuclear (GLE1), regulación del control de la autofagia, la dinámica del citoesqueleto y el metabolismo del ARN (LAS1L, IGHMBP2, AR, SETX), transporte molecular por canalización de cationes (TRPV4), transporte nuclear (LMNA), transcripción nuclear (AR), transporte axonal (BICD2 y DYNC1H1), regulación inmunológica e inflamatoria (TFG), acumulación de gangliósidos en el lisosoma (HEXB), anomalías estructurales y funcionales de las mitocondrias (CHCHD10) 9,15.
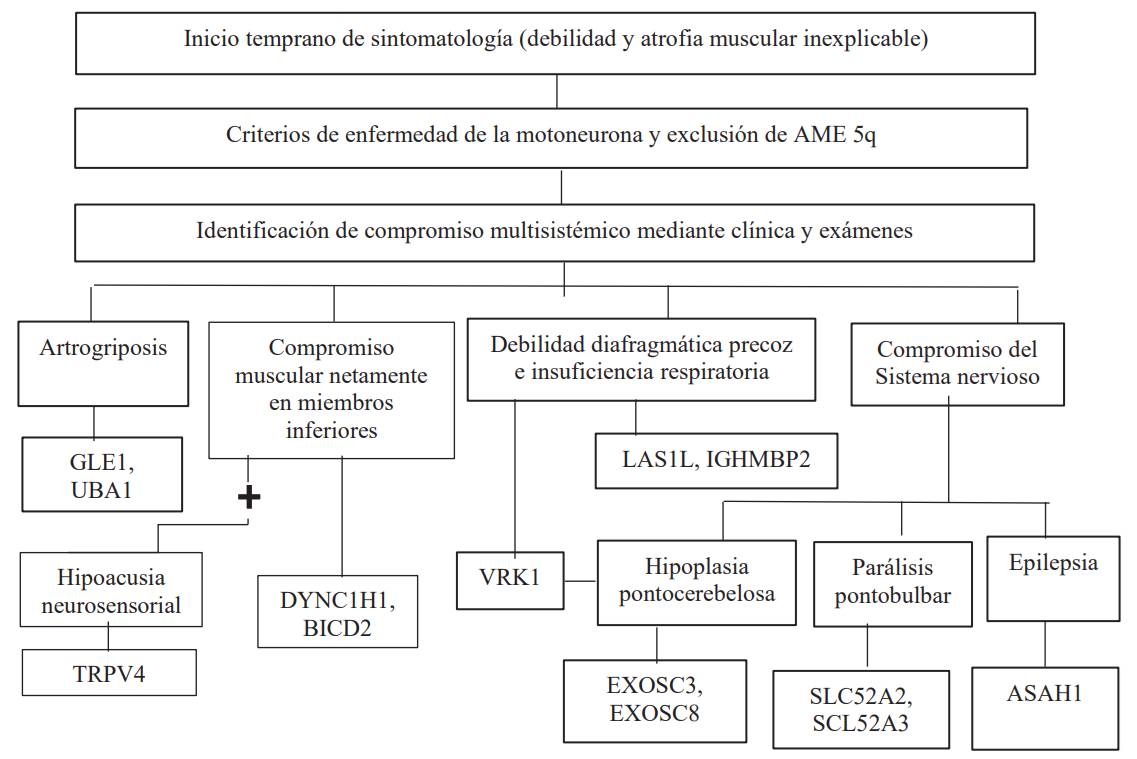
Fuente: Peeters K. Clinical and genetic diversity of SMN1-negative proximal spinal muscular atrophies. Brain. Castiglionia C. Atrofias musculares espinales no asociadas a SMN1. Revista Médica Clínica Las Condes.
Figura 1 Algoritmo de sospecha diagnóstica en AME no 5q de inicio temprano
El espectro clínico de la AME es amplio y heterogéneo, depende de cada variante genética y de las áreas comprometidas. La principal manifestación clínica del compromiso en las motoneuronas inferiores es la debilidad muscular (proximal o distal, simétrica o asimétrica, en miembro superior o inferior y con afectación o no de la musculatura respiratoria). Si existe compromiso de las motoneuronas bulbares, los trastornos de la deglución y la voz estarán presentes. Finalmente, la hiporreflexia o arreflexia, fasciculaciones musculares o linguales, temblor fino de manos orientarán hacia un compromiso de segunda motoneurona.
La presentación clínica de algunos tipos de AME no 5q puede ser similar a la AME 5q en la que se evidencian cuadros de hipomotilidad fetal, debilidad muscular, hipotonía, arreflexia, diplejía facial, artrogriposis o insuficiencia respiratoria; sin embargo, el estudio complementario, el tratamiento y el asesoramiento genético son claramente diferentes 2,4,14,16.
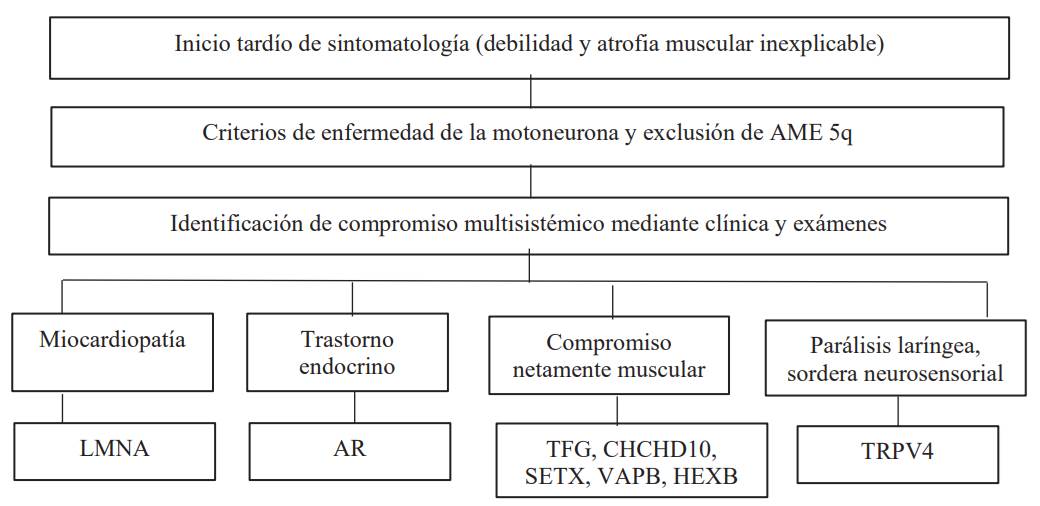
Fuente: Peeters K. Clinical and genetic diversity of SMN1-negative proximal spinal muscular atrophies. Brain. Castiglionia C. Atrofias musculares espinales no asociadas a SMN1. Revista Médica Clínica Las Condes.
Figura 2 Algoritmo de sospecha diagnóstica en AME no 5q de inicio tardio
La AME es considerada un reto médico. La sospecha diagnóstica se inicia ante un paciente con clínica consistente en: debilidad muscular o hipotonía, no atribuida a causas secundarias; signos clínicos que sugieren compromiso de la neurona motora inferior (atrofia muscular, fasciculaciones, hipotonía, pie equino, contracturas deformantes, escápula alada, hipoarreflexia o arreflexia). También se deben tener en cuenta la edad de inicio de las manifestaciones clínicas tipo temprano (congénito, infancia precoz, niñez temprana) o tardío (adulto joven, adulto mayor); la progresión de la sintomatología: rápida o lenta; la afectación de otros sistemas u órganos y el conocimiento de las distintas entidades descritas en las tablas 1 y 2, todo lo cual permitirá una correcta toma de decisiones 2,3.
La confirmación del diagnóstico se realiza mediante pruebas genéticas moleculares que analizan variantes genéticas específicas como la deleción en homocigosis de los exones 7 de SMN1, que es la variante genética más común en la AME. Sin embargo, debido a la amplia variabilidad genética de AME no 5q, la ausencia de variantes patogénicas en SMN1 no descarta la presentación de una AME. La electromiografía y la biopsia muscular formaron anteriormente parte estándar de la evaluación diagnóstica de la AME; no obstante, en la actualidad las pruebas genéticas moleculares están ampliamente disponibles 2,3.
Hoy no existe un tratamiento establecido para cada una de las AME no 5q. Según el Clinical Trials se han documentado desde 2000 hasta el presente año, 215 estudios relacionados con AME, entre los que se encuentran dos estudios asociados a AME no 5q: un ensayo clínico con administración intratecal de genes en fase I/IIa para enfermedades relacionadas con el gen IGHMBP2 con fecha estimada de finalización del estudio en noviembre de 2028; el otro ensayo para evaluación de la seguridad y eficacia de acetato de leuprorelina con 11,25 mg en pacientes con atrofia muscular espinal y bulbar con fecha estimada de finalización el 31 de agosto de 2025.
Por su parte, en la literatura Lee, Termglinchan, Diecke et al. 20 describen un estudio en el que se creó un modelo de Lamin A/C (LMNA)-miocardiopatía dilatada in vitro utilizando cardiomiocitos derivados de células madre pluripotentes inducidas específicas del paciente (iPSC-CM) en los que se evidenció que la inhibición farmacológica y molecular de la vía de señalización del factor de crecimiento derivado de plaquetas (PDGF, por sus siglas en inglés) mejoró los fenotipos de arritmia de iPSC-CM mutantes in vitro que sugiere como un objetivo terapéutico potencial el receptor beta de PDGF; sin embargo, no se han establecido las dosis adecuadas o las alternativas a estos inhibidores por medio de ensayos clínicos; por tanto, el manejo terapéutico de los pacientes con AME no 5q se limita a terapias de apoyo ortopédico, nutricional, ventilatorio y de rehabilitación, según la clínica que presente el paciente 1,17-20.
El seguimiento de los pacientes diagnosticados con AME en etapa presintomática requiere vigilancia del desarrollo de sintomatología, para determinar el inicio adecuado de terapias dirigidas o de apoyo. La evaluación multidisciplinaria se debe hacer cada seis meses para evaluar funciones respiratoria, motora, estado ortopédico y estado nutricional, este apoyo es esencial para reducir la gravedad de los síntomas 23.
Al tratarse de una enfermedad neuromuscular progresiva el pronóstico funcional, la capacidad respiratoria y la expectativa de vida dependerán del tiempo de evolución de la enfermedad, el manejo multidisciplinario de manera oportuna y la instauración o no de complicaciones entre las cuales se encuentran problemas de deglución con posterior afectación en la nutrición; disfunción gastrointestinal con presentación de estreñimiento, reflujo gastroesofágico y retraso en el vaciamiento gástrico y problemas respiratorios consistentes en obstrucción de la vía aérea e infecciones por aspiración. El pronóstico de un paciente con presencia de complicación y sin intervenciones multidisciplinarias tiene una expectativa de vida rara vez mayor a los 2 o 3 años 4,24.
Materiales y métodos
Paciente femenina de 32 años de edad, cuadro clínico de inicio al año y tres meses de vida con equinismo, varismo, supinación del retropié, aducción del antepié derecho y limitación en extensión de la muñeca bilateral, con posterior instauración de debilidad y atrofia muscular en miembros inferiores predominantemente. Niega presencia de alteraciones propioceptivas, auditivas, visuales, cutáneas. Como antecedentes genealógicos: padres no consanguíneos, sin familiares con enfermedades neuromusculares degenerativas.
Al examen físico extremidades simétricas, pulsos simétricos, no frialdad distal, llenado capilar menor de dos segundos, fasciculaciones y dolor a la palpación en el músculo romboides, postura de mano caída bilateral, no hiperextensión, sin hiperlaxitud ligamentaria, con presencia de férulas en miembros inferiores, atrofia muscular, arreflexia generalizada, camina con apoyo, fuerza según escala de Daniels con ausencia de contracción en flexoextensores de manos y pies, signo de Gowers positivo. No logra sedestación, neurológicamente alerta, orientada en tiempo, lugar y persona.
Con paraclínicos en los que se evidencia: creatinquinasa 239 UI/L, calcio 9,4 mg/dl, prolactina 19,14 ng/ml, hormona estimulante de tiroides (TSH) 5,04 mUI/L, hormona paratiroidea (PTH) 51,91 pg/mL, vitamina D 25 ng/ml, 17-OH-progesterona 19,7 UI/l, somatomedina C 181 U/ml, electrocardiograma, ecocardiograma transtorácico y holter sin alteraciones cardiovasculares, ortorradiografía de columna con rotoescoliosis de vértice derecha a nivel del dorso lumbar con ángulo de Cobb de 44 grados, biopsia de nervio sural con pérdida de axones con poca desmielinización, hipertrofia de células de Schwann, sin realización de electromiografía.
De acuerdo con las manifestaciones clínicas neuromusculares degenerativas y resultados de biopsia de nervio sural, la primera impresión diagnóstica fue la enfermedad de Charcot-Marie-Tooth motivo por el cual se realizó estudio molecular de deleciones/duplicaciones en el gen PMP22 del que se obtiene como resultado ausencia de alteración.
Ante la sospecha de una polineuropatía de tipo neurodegenerativa progresiva a clasificar, y teniendo en cuenta la amplia variabilidad fenotípica y genotípica asociados a esta condición médica, se realizó estudio de secuenciación de exoma clínico trío utilizando tecnología Illumina a partir de una muestra de sangre venosa periférica. El ADN se extrajo mediante paquete DNeasy de Qiagen; para determinar su concentración y su pureza, las muestras fueron evaluadas mediante un espectrofotómetro (Na-noDrop), que arrojó valores aproximados de 500 ng/uL y una densidad óptica media (DO) A260/A280 de 1,80. Posterior a esto se realizó secuenciación masiva de librerías Nextera TM mediante plataforma Illumina con cobertura de 100X. Alineamiento con genoma de referencia GRCh38. Todas las regiones seleccionadas presentaron profundidad mayor o igual a 32,2 x y un umbral de confianza mínimo de mapeo Q30 con una lectura total de 27.320.632 bibliotecas Nexteratm Illumina. Los resultados se compilaron en un archivo de salida del tipo Variant Call Format (VCF), en donde se registraron las variantes encontradas. Se consultaron las bases de datos poblacionales Exac, 1000 genomas, OMIM y gnomAD para determinar la existencia de las variantes reportadas; posteriormente se procedió a realizar el análisis bioinformático y de predicción de efecto funcional de las variantes encontradas.
Para el análisis de las variantes reportadas se utilizaron los softwares bioinformáticos Mutation Taster (http://www.mutationtaster.org/), Protein Variation Effect Analyzer (PROVEAN) (http://provean.jcvi.org/index.php), el predictor UMD (http://umd-predictor.eu/), Polyphen (http://genetics.bwh.harvard.edu/pph2/), SIFT (https://sift.bii.a-star.edu.sg/), Human Splicing Finder (http://umd.be/Redirect.html) y Clinvar (https://www.ncbi.nlm.nih.gov/clinvar/), que sirvieron como herramientas in silico de predicción clínica. La nomenclatura utilizada para nombrar las variantes se basó en las recomendaciones del Colegio Americano de Genética y Genómica Médica (ACMG, por sus siglas en inglés). Finalmente se realizó una red de interacción génica por medio del programa GeneMania para determinar asociaciones cercanas con otros genes que permitieran determinar interacciones físicas o niveles de coexpresión.
Resultados
En la secuenciación de exoma clínico trío se encontró una variante patogénica en el gen DYNC2H1 con cambio de nucleótido: c.8365T>C, proteína: p. Phe2789Leu, cigosidad: homocigoto, padre y madre heterocigoto; gen codificante de dineínas citoplasmáticas de la cadena pesada 1, subunidad de la proteína motora primaria responsable del transporte axonal retrógrado en las neuronas que al estar afectado se asocia a la presentación de afectación muscular predominantemente en extremidad inferior proximal 25.
Por otra parte, se encontró la segunda variante con significado clínico patogénico en la secuenciación de exoma clínico trío en el gen NOD2; esta variante no se identificó en el ADN extraído de ninguno de los padres, con cambio de nucleótido: c.1001G>A, proteína: p.Arg334Gln, cigosidad: heterocigoto, gen que desempeña un papel importante en la función del sistema inmunitario; sin embargo, la variante genética no se ha asociado con algún tipo de manifestación clínica según la base de datos ClinVar y el Instituto Europeo de Bioinformática NCBI 27.
Adicionalmente, para determinar interacciones entre los genes afectados y establecer una relación fenotipo-genotipo, se construyó la red de interacción génica (figura 3), en la que se observaron los dos genes en los que se encontraron variantes patogénicas en la paciente: DYNC2H1 y NOD2. Según el análisis con GeneMania presentaban interacciones cercanas con los genes RIPK2, NLRP1, NOD1, IKBKG, DYNC2LI1, NLRC4, DCTN1 y CASP1, todos con funciones relacionadas con procesos al dominio de unión a nucleótidos, vía de señalización del receptor que contiene repeticiones ricas en leucina, vía de señalización del receptor tipo Toll dependiente de MyD88, activación del zimógeno, vía de señalización del receptor 4 tipo Toll, regulación de la secreción de interleucina-1 beta, procesamiento de antígenos y presentación de antígenos peptídicos o polisacáridos por medio de complejo mayor de histocompatibilidad (MCH) clase II. Los genes destacados por su coexpresión con los genes DYNC2H1 y NOD2 fueron RIPK2, NOD1 y TNFAIP3.
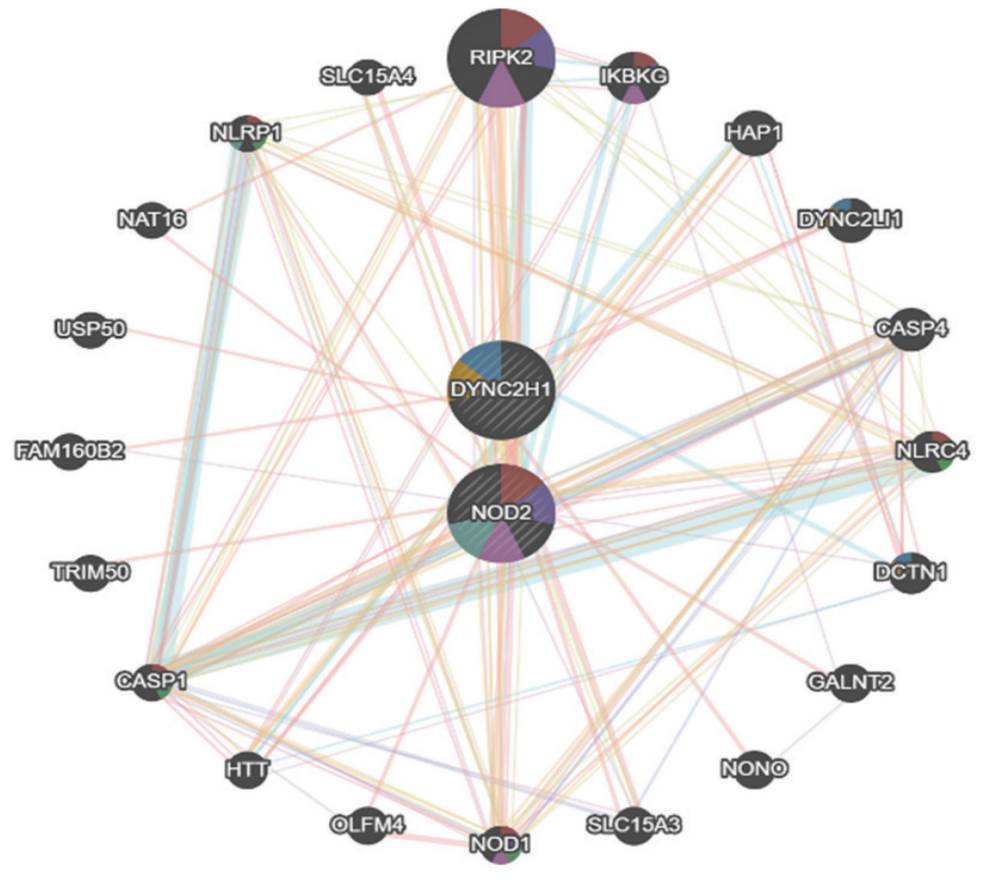
Fuente: elaboración propia, Programa GeneMania.
Figura 3 Red de interacción entre genes DYNC2H1, NOD2 y genes asociados
El gen DYNC2H1 presentó interacción física con seis genes diferentes, teniendo con uno de ellos (DYNC2LI1) una alta interacción. El gen NOD2 por su parte fue el que más interacciones físicas reportó 16. La asociación con el gen RIPK2 fue la más fuerte de ellas.
Conocer las rutas metabólicas y las interacciones permite identificar los mecanismos fisiopatológicos implicados y establecer un diagnóstico específico en una enfermedad neuromuscular degenerativa y progresiva con una variabilidad fenotípica y genotípica como las AME no 5q en aras de ofrecer un tratamiento dirigido que logre impactar en el pronóstico y morbimortalidad atribuida a esta patología, estableciendo pautas de seguimiento, educación sobre riesgo de heredabilidad y una adecuada consejería genética acercándonos a la medicina 4P que impactaría en la historia natural de la enfermedad 2,4,28.
Aspectos bioéticos
El presente reporte de caso se considera un estudio descriptivo observacional basado en la revisión de la historia clínica de un paciente y revisión de la literatura. No se realizó modificación intencionada de las características biológicas, fisiológicas o psicológicas, razón por la cual el nivel de esta investigación se ha categorizado como de riesgo mínimo, según la Resolución 8430 de 1993. Previo a la producción del reporte de caso se realizó el respectivo consentimiento informado para el uso de datos de forma confidencial. Los autores declaran que en este artículo los datos tomados de la historia clínica se analizaron para proteger la confidencialidad y privacidad del paciente.
Discusión
En Colombia las AME se consideran enfermedades huérfanas que han coexistido en la población desde su origen; no obstante, han pasado inadvertidas en relación con su baja prevalencia y desconocimiento del personal de la salud lo que implica que el diagnóstico sea más difícil. Según lo descrito por Peeters et al.9,32 las AME 5q representan el 80 % de los trastornos de la motoneurona, pero la cantidad de genes causales asociados con las AME no 5q, se ha expandido rápidamente debido al advenimiento de las tecnologías de secuenciación de nueva generación. Para Castiglioni y Lozano 15 hasta 2018 existían 20 genes asociados, razón por la cual al obtener un estudio molecular que reporte significado clínico no patogénico en el gen SMN1 no se descarta la enfermedad y se deberán incluir más datos genómicos para la caracterización del paciente con sospecha de AME.
La secuenciación del exoma busca obtener la máxima información genética posible de los exones presentes en los casi 26.000 genes de los seres humanos, cubriendo aproximadamente el 85 % de las variantes asociadas a un heterogéneo grupo de enfermedades genéticas hereditarias complejas, dilucidando el diagnóstico del fenotipo característico del paciente y el descubrimiento de nuevas variantes genéticas asociadas con patologías 28.
Actualmente, como parte del análisis del exoma dirigido a genes de interés, está en uso la secuenciación del llamado exoma clínico, mediante kits desarrollados por la industria como Agilent SureSelect Focused Exome, Illumina TruSight One, Roche NimbleGen SeqCap EZ MedExome, que incluyen unos 5000 genes en el catálogo Online Mendelian Inheritance in Man (ONIM) y asociados a fenotipos clínicamente relevantes relacionados con enfermedad. Es decir, únicamente cubren el 20 % de todo el exoma y requieren, al igual que los paneles de genes, análisis adicionales si no se encuentra la alteración causante de la clínica. El uso clínico se ha propuesto como primera prueba diagnóstica en trastornos del neurodesarrollo 28.
La bioinformática clínica se define como disciplina científica emergente en las ciencias biomédicas que utilizan la tecnología de la información para organizar, analizar y distribuir información biológica, usando ADN, ARN, secuencias de aminoácidos, proteínas, estructuras moleculares tridimensionales, interacciones génicas, vías metabólicas, entre otros; surgió a principios de los noventa a partir de las bases de datos obtenidas de Proyecto Genoma Humano y por medio de la experimentación in silico, lo que ha permitido explicar la función de diversas proteínas, desarrollar modelos estructurales, redes de interacción entre proteínas, dilucidar los diferentes mecanismos moleculares relacionados con la presentación de diferentes enfermedades, mejorar la comprensión de la relación entre la herencia y el riesgo de padecer una afección, revelar la influencia genética en su aparición e identificar biomarcadores y estrategias terapéuticas específicas para estas condiciones 29.
Buscando definir un diagnóstico especifico según las manifestaciones clínicas heterogéneas de la paciente se realizó un estudio de secuenciación de exoma clínico trío, en busca de variantes genómicas asociadas al fenotipo del paciente y realizar una correcta correlación de la función proteica mediante técnicas bioinformáticas. En este contexto, en el presente estudio se identificaron dos variantes genéticas con una significancia clínica patogénica en el gen DYNC1H1 y NOD2 28.
El gen DYNC1H1 codifica la proteína dineína-2, con posterior desarrollo de complejo dineína-2 que se encuentra en los cilios, proyecciones microscópicas que sobresalen de la superficie de las células. La dineína-2 está involucrada en el transporte intraflagelar (TIF) por el cual los materiales se transportan dentro de los cilios; específicamente, la dineína-2 es un motor que utiliza energía de la molécula ATP para impulsar el transporte de materiales desde la punta de los cilios hasta la base 25.
La familia de genes DYNC1H1 y DYNC2H2 codifica dineínas citoplasmáticas de la cadena pesada 1, una subunidad de la proteína motora primaria responsable del transporte axonal retrógrado en las neuronas. Al presentarse una variante genética con significado clínico patogénico en este gen se ven alteradas estas funciones, lo que confirma la causa de AME no 5q en la paciente 25.
Según estudios realizados por Weedon et al.26 que identificaron una mutación sin sentido [p.H306R (c.917A> G)] en el gen DYNC1H1 en una familia de cuatro generaciones con 23 miembros, la característica más llamativa entre los pacientes fue una distribución única de la afectación muscular. El músculo cuádriceps femoral se vio afectado desde el curso temprano de la enfermedad y la extremidad inferior proximal estaba predominantemente involucrada a lo largo del curso de la enfermedad.
Harms et al.25 reportaron tres variantes sin sentido en la cola del dominio de DYNC1H1 en familias con atrofia muscular espinal dominante con predominio de extremidades inferiores (SMA-LED, OMIM 158600), expandiendo el papel de DYNC1H1 al mantenimiento de las neuronas motoras. En su trabajo propusieron que el curso clínico no progresivo de la enfermedad, a pesar del inicio de la primera infancia debería ser otro sello distintivo de SMA-LED; además postuló que la familia de genes asociado a DYNC1H1 desempeña un papel esencial en el mantenimiento de las neuronas motoras espinales y sus axones.
Tsurusaki et al.10 estudiaron dos hermanos: el primero tuvo un retraso motor leve en la infancia, marcha inestable que persistió hasta los tres años, exámenes que mostraban atrofia muscular proximal del miembro inferior y disminución del reflejo tendinoso profundo; el signo de Gowers fue positivo. No se demostró déficit neurológico. Velocidad de conducción nerviosa motora dentro de los límites normales. La tomografía computarizada muscular demostró atrofia severa y degeneración lipídica, predominantemente en el cuádriceps bilateral músculo femoral. Una biopsia muscular del músculo cuádriceps femoral demostró atrofia severa de agrupamiento de fibras tipo 2.
El segundo paciente presentó retraso en el desarrollo motor; el examen físico reveló debilidad muscular moderada en la extremidad inferior proximal, pero el signo de Gowers fue negativo, la tomografía computarizada muscular reveló atrofia severa y degeneración lipídica, principalmente en el cuádriceps femoral bilateral. La debilidad proximal de las extremidades inferiores y el desgaste es evidente, pero el paciente no mostró debilidad en extremidad superior. Los investigadores encontraron variantes sin sentido en los genes DYNC1H1 y DYNC2H2 en los dos pacientes 10.
Por su parte Scoto et al.21 describe el fenotipo de los pacientes con significado clínico patogénico en el gen DYNC1H1 que habitualmente se caracteriza por debilidad muscular con predominio en miembros inferiores en segmentos proximales que preserva relativamente músculos aductores y semitendinosos, atrofia muscular con ausencia de alteración sensitiva, marcha anadina, retraso en la marcha y pérdida de reflejos osteotendinosos distales.
Por otro lado, la variante patogénica en el gen NOD2 no identificada en el ADN extraído de leucocitos de ninguno de los padres, anteriormente conocido como CARD15 según Guzmán et al.22 una proteína que desempeña un papel importante en la función del sistema inmunitario, es activa en algunos tipos de células del sistema inmunitario (incluidos monocitos, macrófagos y células dendríticas), que ayudan a proteger el cuerpo contra bacterias y virus.
La proteína NOD2 tiene varias funciones críticas en la defensa del cuerpo contra los invasores extraños, está involucrada en el reconocimiento de ciertas bacterias y en la estimulación del sistema inmunitario para que responda adecuadamente. Cuando se activa por sustancias específicas producidas por bacterias, la proteína NOD2 activa un complejo proteico llamado factor nuclear-kappa-B, que regula la actividad de múltiples genes, incluidos los que controlan las respuestas inmunes y las reacciones inflamatorias. No obstante, a pesar de presentar variantes genéticas no se asocia con ningún tipo de manifestación clínica 27.
Conclusión
La AME no 5q es un conjunto de trastornos neurodegenerativos hereditarios capaces de causar una alteración en las células del asta anterior en la médula espinal y los núcleos motores en la parte inferior del tronco encefálico y posteriormente una alta variabilidad fenotípica y genotípica, que generan un impacto sobre la calidad de vida, desarrollo psicosocial, emocional y funcional de las personas.
Aunque en la mayoría de los pacientes se debe a variantes en el gen SMN1 existen otros genes no 5q asociados a esta patología, razón por la cual surge la importancia de realizar estudios genómicos, la bioinformática clínica, las redes de interacción y el fenotipado inverso basado en la selección de un grupo de personas con una variante genética y la evaluación de su fenotipo, los cuales se han convertido en herramientas fundamentales para la caracterización de estas enfermedades complejas y la determinación con potencial de cambiar la medicina reactiva en medicina preventiva.
De este modo se logra realizar un diagnóstico precoz, con la posibilidad de iniciar un tratamiento temprano y dirigido que impacte en el pronóstico y morbimortalidad atribuida a esta patología, estableciendo pautas de seguimiento y una adecuada consejería genética que permita educación sobre riesgo de heredabilidad o presentación de una enfermedad acercándonos a la medicina de precisión en aras de la medicina de las 4P que impactarían en la historia natural de la enfermedad 1,15,31.

