










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkCiencia en Desarrollo
Print version ISSN 0121-7488
Ciencia en Desarrollo vol.5 no.2 Tunja July/Dec. 2014
Aplicación de métodos compuestos en la estimación de las entalpías de formación de compuestos peróxidos ROOR' (R, R'= H, F, Cl y NO)
Applying Composite Methods for Estimating Enthalpies of Formation of Peroxides ROOR' (R, R'= H, F, Cl y NO)
C. Buendía-Atencioa,*
R. Vivas-Reyesb
J. A. Gómez Castañoc
V. P. Lorett Velásquezd
a Grupo Sistemas Complejos, Facultad de Ciencias Básicas, Departamento de Química, Universidad Antonio Nariño, Sede Circunvalar, Bogotá D. C., Colombia.
* Autor de correspondencia: c.buendia@uan.edu.co
b Grupo de Química Cuántica y Teórica, Facultad de Ciencias Exactas y Naturales, Universidad de Cartagena, Cartagena D. T y C., Bolívar, Colombia.
c Laboratorio de Química Teórica y Computacional, Grupo de Investigación en Química-Física Molecular y Modelamiento Computacional (QUIMOL), Escuela de Ciencias Químicas, Facultad de Ciencias, Universidad Pedagógica y Tecnológica de Colombia (Uptc), Tunja, Boyacá, Colombia.
d Universidad de Cundinamarca, Facultad de Ciencias Agropecuarias, Sede Facatativá, Cundinamarca, Colombia.
Recepción: 04-mar-14 Aceptación: 13-oct-14
Resumen
Diversos métodos compuestos de alto nivel en química cuántica, tales como las teorías Gaussian-n y Weizmann-n, han sido empleados para estimar las entalpías de formación de contaminantes atmosféricos peroxídicos a partir de energías de atomización totales y de esquemas de reacciones isodésmicas. Los valores estimados para la ΔHof,298K de HOOH, HOOF, HOOCl, HOONO, FOOF, FOOCl, FOONO, ClOOCl y ClOONO son −32,2 ± 0,8; −10,9 ± 1,1; −8,4x10 − 2 ± 1,2; −2,9 ± 0,7; 8,4 ± 1,6; 21,5 ± 1,3; 19,7 ± 1,3; 32,3 ± 1,4y −31,3 ± 0,8 kcal mol−1, respectivamente. Una comparación entre los valores experimentales existentes y los valores teóricos obtenidos es presentada en este trabajo, destacando la alta precisión química con un bajo costo computacional.
Palabras clave: Entalpía de formación, Energías de atomización, Métodos compuestos, Peróxidos, Reacciones isodésmicas.
Abstract
Different high-level quantum chemical composite methods such as the Gaussian-n and Weizmann-n theories have been used to derive enthalpy of formation values from calculated atomization energies and scheme isodesmic reactions of peroxide pollutants. The resulting values of ΔHof,298K de HOOH, HOOF, HOOCl, HOONO, FOOF, FOOCl, FOONO, ClOOCl y ClOONO son −32,2 ± 0,8; −10,9 ± 1,1; −8,4x10 − 2 ± 1,2; −2,9 ± 0,7; 8,4 ± 1,6; 21,5 ± 1,3; 19,7 ± 1,3; 32,3 ± 1,4 and −31,3 ± 0,8 kcal mol−1, respectively. A comparison with previous experimental and theoretical values are presented highlighting the high chemical accuracy with low computational cost.
Key words: Atomization Energies, Composited Methods, Enthalpy of Formation, Isodesmic Reactions, Peroxides.
1. Introducción
En la actualidad, el desarrollo de las tecnologías informáticas se mueve a un ritmo incesante, gracias, principalmente, a la implementación de nuevos materiales optimizados sobre interfaces de alto rendimiento en los equipos de cómputo modernos. En el campo de la química computacional tales avances han permitido gradualmente ir disminuyendo el elevado costo computacional requerido para la ejecución de cálculos mecano-cuánticos de alto nivel por los métodos QCISD(T), CCSD(T), CISDTQ, etc., combinados con grandes conjuntos de bases, lográndose así el alcance de una precisión química de ∼ 1 kcal mol−1 en la determinación de diversas propiedades energéticas moleculares. Si bien el acceso a cálculos refinados ha aumentado considerablemente en los últimos años, la inversión financiera necesaria para la adquisición de los avanzados sistemas de cómputo utilizados para su ejecución continúa siendo un impedimento difícil de superar por centros académicos y grupos de investigación pequeños, con recursos limitados.
Para superar dicha dificultad se han desarrollado e implementado métodos conformados por una serie de cálculos con diferentes funciones de base y niveles de teoría, denominados métodos compuestos, los cuales permiten obtener resultados muy próximos a los generados a partir de un cálculo de alto nivel, pero con un menor costo computacional [1]. Entre los métodos compuestos más utilizados se encuentran la serie de base completa (CBS), las teorías Gaussian-n y los métodos ab initio Weizmann-n (Wn,n = 1 − 4).
Los métodos CBS [2] involucran esencialmente siete u ocho pasos, a saber: a) una optimización de la geometría (a un nivel HF/3-21G* o MP2/6-31G*, dependiendo del método CBS en particular); b) un cálculo de las energías vibracionales del punto cero (ZPE, por su sigla en inglés -Zero Point Energy-); c) un cálculo del punto simple HF con una gran base, por ejemplo, suelen utilizarse las bases 6 − 311 + G(3d2 f,2df, p)o6 − 311 +G(3d2 f,2df,2p), cuya selección depende del método CBS utilizado; d) un cálculo de energía al nivel MP2 con un conjunto de base, cuya elección dependerá nuevamente del método CBS empleado; e) la extrapolación orbital del par natural, la cual estima el error debido al uso de un conjunto de base finito; f) un cálculo de energía al nivel MP4; g) un cálculo de energía QCISD(T), para algunos métodos CBS, y, por último, h) una o más correcciones empíricas.
En los métodos compuestos Gn se adoptan también correcciones empíricas; por mencionar uno de ellos, en el método G3, el término E(HLC) es la corrección de alto nivel cuyo propósito es corregir el truncamiento de la base, y está dada por (1).
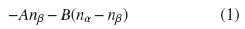
donde nα y nβ son el número α y β de electrones de valencia, con nα ≥ nβ, y los términos A y B son parámetros empíricos que equivalen para moléculas A = 6,386x10−4, B = 2,977x10−4 y para átomos A = 6,2194x10−4, B = 1,185x10−4 hartrees, respectivamente [3].
La expresión total para el cálculo de la energía del método compuesto G3 es:

donde la energía E(ZPE) y las frecuencias de las estructura de partida son obtenidas a un nivel HF/6 − 31G(d) y escaladas por un factor de 0,893. El término E(SO) corresponde a la corrección de espínorbita, y el nivel teórico MP2(FU) implica todas las posibles excitaciones.
La teoría G4, una formulación Gn recientemente desarrollada por Curtiss et al. [4], presenta una desviación media absoluta de 0,83 kcal mol−1 en la estimación de 454 energías compuestas por entalpías de formación, potenciales de ionización, afinidades electrónicas, protónicas y energías de enlaces de hidrógeno. Las modificaciones incluidas en este método son: a) un procedimiento de extrapolación obtenido a partir de un cálculo Hartree-Fock limite (HF) para su inclusión en el cálculo de la energía total; b) un incremento en el conjunto de polarización de d a 3d sobre los átomos de la primera fila y 4d sobre los átomos de la segunda fila con reoptimización de los exponentes del conjunto para 4d; c) el reemplazo del método QCISD(T) por CCSD(T); d) la obtención de las geometrías y la energía del punto cero al nivel de teoría B3LYP/6 − 31G(2df, p), y, por último, e) dos nuevos parámetros para la corrección de alto nivel (HLC) que tienen en cuenta las deficiencias de los radicales y de especies con un solo par de electrones en la capa de valencia. La expresión para el cálculo de la energía del método G4 es presentada en (3).
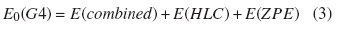
donde el término E(combined) viene definido por (4):

Por su parte, los métodos compuestos Weizmannn, W1 y W2, desarrollados por Martin y De Oliveira [5-6], mostraron un error medio absoluto de 0,37 kcal mol−1 y 0,23 kcal mol−1, respectivamente, en la estimación de la energía de atomización molecular de un conjunto de datos experimentales de 28 moléculas. Este nivel de precisión es alcanzado gracias, principalmente, a la correlación consistente ofrecida por los conjuntos de bases desarrollados por Dunning y colaboradores [7-8], los cuales son utilizados ampliamente a lo largo de la metodología; por ejemplo, en los métodos compuestos W1 y W2 la optimización de la geometría es calculada inicialmente a los niveles de teoría B3LYP/VTZ +1 y CCS D(T )/VQZ +1, respectivamente [9].
Particularmente, uno de los sistemas químicos que ha sido estudiado teóricamente a altos niveles, debido a su "simple" complejidad molecular e importancia en áreas de investigación como biología, química preparativa, química de la combustión, procesos farmacéuticos y medio ambiente, son los peróxidos formados por 4 y 5 átomos, ROOR' [10-12]; la presencia del enlace O-O en su estructura molecular es la causa principal de su marcada actividad química en procesos de oxidación y radicalarios. Entre estos sistemas, el miembro más sencillo corresponde al peróxido de hidrógeno, HOOH, que es usado a multiescala a nivel mundial en procesos industriales, por su bajo costo y fácil disponibilidad de sus oxígenos [11]. A nivel ambiental los peróxidos son considerados contaminantes atmosféricos por su alta reactividad con especies radicalarias en la generación y consumo de ozono a través de procesos fotoquímicos en la troposfera y estratosfera [12]. Dependiendo de los grupos sustituyentes R y R' se derivan nuevas familias de compuestos, tales como los peróxidos de alquilo ROOR' (RR'=CnH2n+1), halo peróxidos XOOX (X= halógenos e hidrogeno) [13], peroxinitratos de alquilo ROONO2 [14-16] y acetil peroxinitratos de alquilo (PAN) R(C=O)OONO2 [17]
Tal como se ilustra en la figura 1, la estructura molecular de los compuestos peroxídicos de cadena abierta presentan niveles de torsión variables alrededor del enlace O-O, originando diferentes isómeros estructurales.
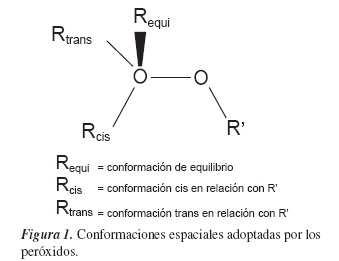
En este sentido, las estructuras de equilibrio y propiedades energéticas de los distintos isómeros de los compuestos HOOF, HOOCl, HOONO, HOOCN, FOOF, ClOOF, ClOOCl y FOONO fueron estudiadas recientemente a través de cálculos químico-cuánticos a los niveles de teoría B3LYP/6311++G(3df,3pd) y CCSD(T)/aug-cc-pVDZ, lográndose una excelente concordancia con los valores experimentales disponibles [18-19].
En otros estudios, J. S. Francisco [20] estimó una entalpía de formación de 0,4 ± 2 kcal mol−1 para HOOF, empleando la Teoría de Perturbaciones a Cuarto Orden de Møller-Plesset (MP4). Posteriormente, el mismo autor reexaminó esta propiedad al nivel teórico QCISD(T)/6-311++G(2df,2p), obteniendo un nuevo valor de −9,3 ± 2 kcal mol−1, que resultó consistente con el derivado a partir de la reacción de descomposición HOOF → HO + FO de −8,4 ± 5 kcal mol−1, en la cual se consideraron valores de ΔHdiss = 45,4; ΔHf,OH = 9,2y ΔHf,FO = 27,8 kcal mol−1 [21]. Tal discrepancia motivó la reexaminación de la entalpía de otros peróxidos de la serie. Por ejemplo, Lee y Rendell [22] estimaron una ΔH◦f,298K = 0,2 kcal mol−1 para el HOOCl, empleando métodos basados en la teoría de coupled clusters (CCSD(T)), estudio dentro del cual fueron empleadas las energías moleculares calculadas y los valores experimentales de la entalpía de formación de los radicales HO• y ClO• para predecir una energía de disociación de enlace (BDE) de la ruptura homolítica O-O de 33,4 kcal mol−1, que es muy cercano al obtenido por Weissman y colaboradores, de 33,5 kcal mol−1, a partir de diversas metodologías cinéticas [23].
Las entalpías de formación de algunos peróxidos ROOR' fueron estudiadas por Dixon y colaboradores [24-26] a altos niveles de teoría, empleando métodos basados en la teoría de coupled clusters CCSDT, CCSDTQ y CCSDTQ5 en combinación con los extensos conjuntos de base CBS y aug-cc-pVnZ (con n = D,T,Q,5,6 y 7), obteniendo un error medio absoluto de 0,1 kcal mol−1. Igualmente, investigaron la termoquímica de los derivados nitro peróxidos, obteniendo un valor ΔH◦f,298K = −3,0 kcal mol−1 para el HOONO, que difiere significativamente del estimado por McGrath y Roland, de ΔH◦f,298K = −5,7 kcal mol−1, empleando el método compuesto G2 [27].
En razón a las diferencias numéricas reportadas en las entalpías de formación calculadas para los compuestos ROOR' (R, R' = H, F, Cl y NO), en el presente trabajo se presenta una exhaustiva revisión de la termoquímica de los peróxidos mencionados, empleando diferentes técnicas en termoquímica computacional a partir de diversos métodos ab initio de última generación. Los valores obtenidos son comparados con los valores experimentales y teóricos normalmente aceptados en la literatura.
2. Metodología computacional
Todos los cálculos químico-cuánticos fueron realizados con el software Gaussian 09 [28]. Las geometrías moleculares de los compuestos peroxídicos HOOH, HOOF, HOOCl, HOONO, FOOF, FOOCl, FOONO, ClOOCl y ClOONO fueron optimizadas empleando métodos de gradientes analíticos a través del funcional B3LYP y el conjunto de base 631G(2df, p). Para la estimación de las entalpías de formación ΔH◦f,298K fueron empleadas dos metodologías en termoquímica computacional: a) energías de atomización total, ΣD0(M), y b) esquemas de reacciones isodésmicas. En la primera de ellas, la energía de atomización total, definida como la energía necesaria para disociar totalmente una molécula en todos sus átomos constituyentes, fue calculada empleando la ecuación (5).
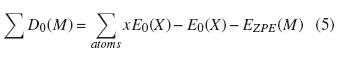
donde E0(X)y E0(M) son las energías de los átomos constituyentes (X) y de la molécula (M), respectivamente, y EZPE(M) es la energía escalada del punto cero de la molécula. El término x representa el número de veces que participa un átomo en particular en la constitución de la molécula. Durante este procedimiento, el resultado de ΣD0(M) fue utilizado para obtener las entalpías de formación a 0 K y a 298 K mediante la ecuación (6).
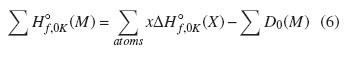
donde ΔH◦f,0K (X) son las entalpías de formación experimental a 0 K de los átomos [29]. En el caso, los valores usados para los átomos H, O, N, F y Cl fueron 51,63 ± 0,00; 58,99 ± 0,02; 112,53 ± 0,02; 18,47 ± 0,07, y 28,59 ± 0,00 kcal mol−1, respectivamente [15].
Posteriormente, para derivar las entalpías de formación a 298 K, ΔH0f,298K , fueron adicionadas las respectivas correcciones térmicas, según se muestra en la ecuación (7).
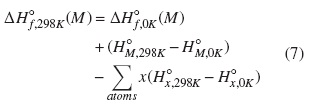
Las correcciones térmicas empleadas para los átomos H, N, O, F y Cl fueron 1,01; 1,04; 1,04; 1,05, y 1,10 kcal mol−1, respectivamente [30]. Respecto a la estrategia de esquemas de reacciones isodésmicas definida como reacciones reales o hipotéticas, en donde el número total de cada tipo de enlace es idéntico en reactivos y productos [31], esta produce datos termoquímicos muy precisos debido a la cancelación de errores sistemáticos inherentes del método de cálculo empleado [32]. Este procedimiento incluye calcular la entalpía de reacción teórica (ΔHr) para la reacción propuesta, continuando con el cálculo de la entalpía de formación (ΔHf ) de la especie de interés a partir de la misma reacción con la utilización de valores experimentales de las otras especies y el ΔHr de reacción teórico obtenido inicialmente. Por ejemplo, para la reacción hipotética A +B → C +D, tenemos que su entalpia de reacción teórica a 298 K, ΔH◦f,298K , está dada por la ecuación (8).
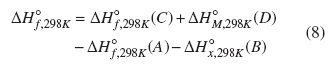
Los valores recomendados para las entalpías de formación han sido calculados a través del valor promedio de los resultados estimados por los métodos compuestos G3B3, G3MP2B3, [33], G4 [4], G4MP2 [34], CBS-QB3 [35] y W1U [5], los cuales tienen una desviación absoluta promedio de 1,13; 1,25; 0,83; 1,04; 1,08 y 0,37 kcal mol−1, respectivamente, por comparación con valores experimentales de un numeroso grupo de compuestos.
3. Resultados y discusión
3.1. Estructuras de equilibrio de los compuestos ROOR'
Los sistemas investigados en este estudio son compuestos derivados del peróxido de hidrógeno (HOOH), en donde uno o los dos átomos de hidrógeno han sido sustituidos por F, Cl y NO. La obtención de las estructuras de equilibrio es el punto de partida para el estudio de la energética de estos compuestos. En general, todos los compuestos peroxídicos estudiados presentaron una estructura semiplanar conformada por el enlace O-O y uno de los grupos sustituyentes R o R'. Los compuestos HOOH, FOOF y ClOOCl han sido estudiados y caracterizados experimentalmente [36-38].
En la figura 2 se presentan las conformaciones de equilibrio obtenidas al nivel B3LYP/6-31G(2df, p) y visualizadas con el programa MOLDEN 5.0 [39].
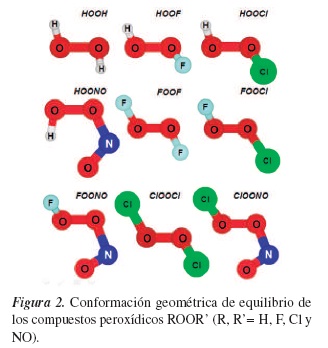
En la tabla 1 se observa que nuestros resultados para los parámetros geométricos de los compuestos HOOH, FOOF y ClOOCl, obtenidos al nivel B3LYP/6-31G(2df, p), están en excelente acuerdo con los datos experimentales y teóricos reportados al nivel de teoría B3LYP/6-311++G(3df,3pd) y CCSD(T)/aug-cc-pVDZ [18] Nuestros resultados para los compuestos HOOF, HOOCl, HOONO, FOOCl, FOONO y ClOONO son presentados en la tabla 2.
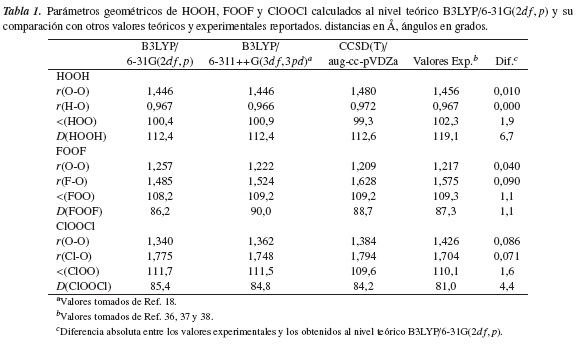
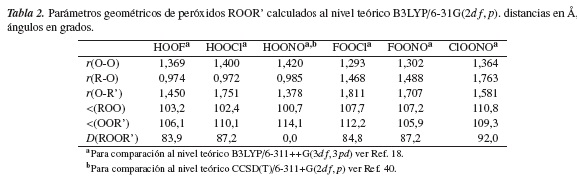
Los resultados encontrados en este estudio al nivel de teoría B3LYP/6-31G(2df, p) para los parámetros geométricos de los compuestos peroxídicos derivados HOOF, HOOCl, FOOCl, FOONO y ClOONO predicen una estructura de equilibrio muy similar a las reportadas previamente al nivel de teoría B3LYP/6-311++G(3df,3pd) y CCSD(T)/aug-ccpVDZ [18]. Por otro lado, la estructura de equilibrio más estable, calculada aquí para el compuesto HOONO, corresponde a una conformación cis plana con un ángulo diedro D(HOON) de 0,0°; un resultado contrario a la estructura de equilibrio semiplana OO-NO con un ángulo diedro D(HOON) de 99,6°, reportada por Maciel y colaboradores [18]. No obstante, nuestra geometría de equilibrio cis-HOONO coincide con la conformación de energía mínima estimada para este compuesto por otros autores [40] al nivel de teoría CCSD(T)/6311+G(2df, p)//B3LYP/6-311+G(d, p). Igualmente, McGrath y Roland [27] identificaron la estructura cis para el compuesto HOONO al nivel de teoría MP2/6311+G(3df,2p) como el confórmero más estable y de menor energía en sus investigaciones.
3.2. Entalpías de formación de los compuestos ROOR' a partir de energías de atomización
En la tabla 3 se presentan las entalpías de formación de los peróxidos ROOR' (R, R'= H, F, Cl y NO) calculadas a partir de las energías de atomización y usando seis diferentes métodos compuestos. Entre estos últimos es destacable la alta precisión de las cuatro teorías Gn(G3B3, G3MP2B3, G4, G4MP2), mediante las cuales se logran entalpías que difieren en menos de 1,1 kcal mol−1 respecto a la mejor magnitud reportada para seis de las nueve moléculas estudiadas: HOOH, HOOF, HOOCl, HOONO, FOOCl y ClOONO. Para los peróxidos halógenados FOOF, FOONO y ClOOCl, las diferencias alcanzadas con los métodos Gn se encuentran entre el rango de 1,6 y 5,5 kcal mol−1. De las cuatro teorías Gn utilizadas, el método más preciso para el cálculo de entalpías de peróxidos ROOR' corresponde al G4, cuyo promedio entre valores absolutos de entalpías para las nueve moléculas es de 1,38 kcal mol−1. Similarmente, el método compuesto Weizmann W1U presenta una precisión elevada en este trabajo, verificable a través de una diferencia igual o menor a 1,1 kcal mol−1 entre las entalpías calculadas con este método y las reportadas para las moléculas HOOH, HOONO, FOOCl, ClOOCl y ClOONO; en promedio, las diferencias absolutas entre todas las moléculas es de tan solo 1,27 kcal mol−1. Mientras tanto, la menor precisión alcanzada fue mediante la serie de base completa CBS-QB3, cuyos valores varían entre 1,2 y 6,4 kcal mol−1 con respecto a los reportados para las moléculas HOOH, HOOF, HOOCl, FOOCl, FOONO y ClOONO. No obstante, es el único método de toda la serie con que se obtiene un valor preciso para la molécula FOOF (con una diferencia de 0,7 kcal mol−1 respecto al reportado), muy lejano a los valores por encima de 3,8 kcal mol−1 calculados con los demás métodos.
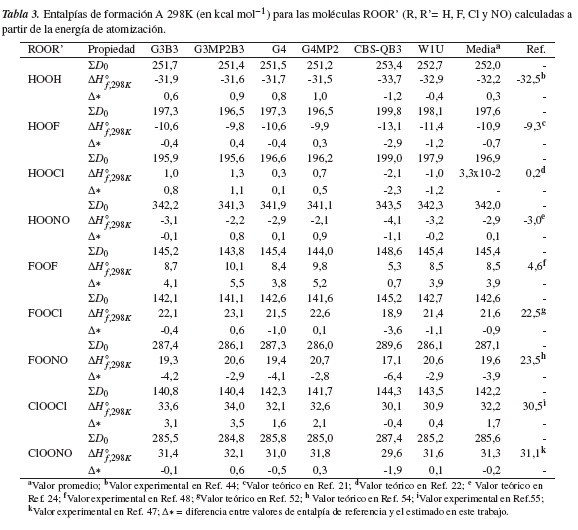
Como puede observarse, los métodos compuestos empleados para estimar entalpías de formación a través de energías de atomización proveen resultados de alta fiabilidad en la mayoría de los casos, y por este motivo han sido empleados en las más recientes publicaciones de nuestro grupo [41-43].
Una forma de evidenciar la confiabilidad de los métodos compuestos usando energías de atomización es a través de los valores promedios (Media), (ver tabla 3). Por ejemplo, el valor medio de entalpía estimado aquí para el peróxido HOOH es de -32,2±0,9 kcal mol−1, muy próximo al dato experimental reportado de -32,5 kcal mol−1 [44]. Este mismo valor medio difiere en menos de 1,0 kcal mol−1 en relación con los resultados encontrados por otros autores (-32,0±0,3 kcal mol−1) empleando altos recursos computacionales y niveles de cálculo CCSDTQ5/aug-cc-pwCVnZ (n = T , Q y 5) [26].
Tal y como se mencionó anteriormente, la entalpía de formación para el HOOF derivada por J. S. Francisco al nivel teórico QCISD(T)/6-311++G(2df,2p) fue de -9,3 kcal mol−1[21], un resultado comparable con los hallados en este trabajo, de -9,8 y -9,9 kcal mol−1, con los métodos G3MP2B3 y G4MP2, respectivamente (ver tabla 3). Los otros métodos compuestos empleados sugieren que la entalpía de formación para el HOOF es ligeramente mayor, razón por la cual su valor promedio es -10,9±1,2 kcal mol−1.
Por su lado, la ΔH◦f,298K recomendada para el peróxido HOOCl es 0,2±1.0 kcal mol−1, de acuerdo con el nivel teórico CCSD(T)/TZ2P usado por Lee y Rendell [22], una cantidad sobreestimada en comparación con nuestro valor promedio de 3,3x10-2±1,3 kcal mol−1.
Para los peroxonitrilos HOONO y ClOONO, los promedios calculados de entalpías en este trabajo son de -2,9±0,7 y 31,3±0,9 kcal mol−1, respectivamente. Tales magnitudes difieren significativamente de las reportadas por McGrath y Rowland [27], quienes calcularon valores de -5,7 (HOONO) y 24,3 (ClOONO) kcal mol−1, usando el método compuesto G2 (desviación media absoluta de 0,92 kcal mol−1). En cambio, nuestros resultados para el compuesto HOONO concuerdan excelentemente con el reportado en los recientes trabajos de Dixon y colaboradores [24], de -3,0 kcal mol−1, empleando las metodologías basadas en la teoría de coupled clusters CCSDT, CCSDTQ y CCSDTQ5 en combinación con extensos conjuntos de base. Por su parte, Colussi y Grela [45] calcularon una entalpía de formación para el ClOONO a 300 K de 35,5±2,0 kcal mol−1, empleando esquemas de aditividad de enlace, aunque en un subsecuente trabajo el nuevo valor reportado por los mismos autores fue de 32,3±3,0 kcal mol−1 [46]. Más recientemente, Szakács y colaboradores [47], empleando el nivel teórico CCSD(T)/cc-pCVTZ, obtuvieron un valor para la entalpia de formación del peroxinitrito de cloro de 31,1±0,7 kcal mol−1, muy cercano a nuestro valor promedio de 31,3±0,9 kcal mol−1.
La entalpía de formación experimental del FOOF fue obtenida por Kirshenbaum en 1959, quien reportó un valor de 4,6±0,5 kcal mol−1 [48]. Estudios teóricos realizados sobre el FOOF tienden a estimar valores más altos. Por ejemplo, Feller y Dixon [25] consiguieron un resultado de 9,6 ± 0,9 kcal mol−1 implementando métodos basados en la teoría de coupled clusters con extensos conjuntos de bases, la cual ofrece un error absoluto de 1,0 kcal mol−1 en la determinación de la entalpía de formación; entre tanto, Jursic halló un valor igual a 7,1 kcal mol−1 usando la técnica de energía de atomización con los métodos compuestos G1, G2 y CBS-Q [52]. Andersson y Roos implementaron métodos multiconfiguracionales CASSCF/CASPT2 y estimaronla energía de atomización para el FOOF de ΣD0 = 136,5 kcal mol−1, de la cual derivaron una entalpía mayor a la experimental reportada [50]. Nuestra ΔH◦f,298K promedio estimada para el FOOF es de 8,5±1,7 kcal mol−1, un resultado más próximo al valor experimental que el reportado por Feller y Dixon. Particularmente, la entalpía de formación para el FOOF, derivada aquí por el método CBS-QB3, de 5,3 kcal mol−1, es comparable con la entalpía experimental. Lo anterior es consistente con la mejor aproximación teórica reportada hasta el momento al nivel de teoría CBS-Q de 7,1 kcal mol−1 [49]. Jursic justifica en su trabajo que la configuración de este método compuesto favorece el estudio de pequeñas moléculas polares. Otros autores [57-58] que han investigado al FOOF han podido concluir que los enlaces F-O hacen parte del grupo de especies de efectos de correlación no-dinámica severa (NDC), ya que poseen un largo número de electrones desapareados que causan repulsiones entre los pares libres del sistema, haciendo difícil la estimación de la geometría molecular. Debido a estos efectos relativistas, que no han sido superados por métodos basados en la teoría de coupled clusters, la determinación experimental realizada en los años sesenta, a partir de una bomba colorimétrica para la reacción F2O2(l) → O2(g) + F2(g) a 190 K medida por Kirshenbaum, seguirá siendo referencia para este peróxido.
Nuestro valor promedio tiene una desviación estándar de 1,9 kcal mol−1, que es relativamente alta; esto es una consecuencia directa si se considera la entalpía de formación derivada con el método CBSQB3.
En cuanto al compuesto FOOCl, J. S. Francisco calculó inicialmente su entalpía de formación de 36,8 kcal mol−1 a un nivel de teoría QCISD(T)/6311G(2df,2p)//MP2/6-311G(2df,2p) [51]. Después, el mismo autor reexaminó este trabajo incrementando el nivel teórico a CCSD(T)/6311++G(3df,3pd)//CCSD(T)/6-311G(2d,2p) y G2, obteniendo entalpías menores de 22,5 y 19,1 kcal mol−1, respectivamente [52]. Nuestra ΔH◦f,298K promedio para el FOOCl es de 21,6±1,5 kcal mol−1, un resultado consistente con los reportados por Jursic [53] y Dibble [54], de 22,8 y 21,4 kcal mol−1 a los nivel de teoría CCSD(T) y B3LYP/6311++G(3df,3pd), respectivamente.
Dibble y Francisco también estimaron la entalpía de formación para el FOONO en 23,5±4 kcal mol−1 a 0 K, usando la teoría de interacción de configuraciones cuadrática QCISD(T) combinada con los conjuntos de bases 6-311G(3df ) y 6-311G(2d) [54]. Nuestro valor promedio obtenido deΔH◦f,298K = 19,6±1,3 kcal mol−1 para este compuesto sugiere una entalpía ligeramente inferior; sin embargo, la alta incertidumbre asociada de 4,0 kcal mol−1 reportada por Dibble y Francisco [54] hace que nuestro valor sea aceptable para la entalpía de formación del FOONO a este nivel. Por último, con la aproximación de las energías de atomización para la entalpía de formación teórica del compuesto ClOOCl obtuvimos un valor promedio de 32,2±0,9 kcal mol−1, el cual está en excelente acuerdo con el valor experimental aceptado de 30,5 ± 0,7 kcal mol−1[55].
3.3. Entalpías de formación de los compuestos ROOR' a partir de energías de esquemas de reacciones isodésmicas
En la tabla 4 son presentadas las entalpías de reacción, ΔH◦r,298K, y de formación, ΔH◦f,298K , de los compuestos ROOR' (R, R'= H, F, Cl y NO), empleando reacciones isodésmicas a los niveles teóricos G4 y G4MP2. Este último incorpora el método CCSD(T) para un nivel más alto de tratamiento de correlación electrónica. Las entalpías de formación para las moléculas HOONO2, H2O, HNO3, HOF, HOCl, CH3ONO, CH3OH, F2O, CF3OF, CF3OH y Cl2O fueron tomadas de la compilación de datos del NIST (National Institute of Standard and Technology) y de la NASA [15,56].

La reacción isodésmica para el HOOH deriva un valor de ΔH◦f,298K = -32,2 kcal mol−1, siendo solamente 0,3 kcal mol−1 menor que el valor experimental reportado [44]. Para el compuesto ClOOCl, las entalpías de formación calculadas a partir de reacciones isodésmicas fueron de 32,5 (G4) y 32,7 (G4MP2) kcal mol−1, que son ligeramente mayores respecto al valor experimental de 30,5 kcal mol−1 reportado para esta especie [55]. Dichos resultados pueden ser atribuidos a una posible acumulación de errores en las medidas experimentales de las entalpías de formación de las otras moléculas de la reacción planteada, por ejemplo, el H2O aporta ± 9,6 x10−3 kcal mol−1 de error, mientras que para el HOONO2 y HNO3 los errores no se han determinado (ver referencias 15, 56).
La reacción isodésmica presentada por Francisco [21] para el HOOF al nivel teórico QCISD(T)/6311++G(2df,2p) produce valores de ΔHr◦ ,0K = -15,4 y ΔH◦f,0K = -9,3±2,0 kcal mol−1, los cuales son comparables con nuestros resultados reportados, teniendo en cuenta el parámetro de corrección térmica de 0 K a 298 K de aproximadamente 1,0 kcal mol−1 (ver tabla 4). Del mismo modo, su resultado para la ΔH◦ f,0K del FOOCl, empleando la reacción isodésmica HOF + HOCl → FOOCl + 2H2O, produce un valor de 22,5 kcal mol−1 al nivel de teoría CCSD(T)/6-311++G(3df,3pd) [52], que es comparable con los estimados en este trabajo, de 21,0 y 21,3 kcal mol−1, a partir de una reacción isodésmica diferente (ver tabla 4).
El valor experimental de la entalpía de formación de 4,6 kcal mol−1 para el compuesto FOOF [48] continúa siendo un dato difícil de reproducir a partir de cálculos computacionales; en nuestro caso, los resultados estimados usando reacciones isodésmicas fueron de 7,8 y 8,1 kcal mol−1 a los niveles G4 y G4MP2, respectivamente. Estos valores son comparables con el valor promedio de 8,5 kcal mol−1 obtenido por atomización de las energías totales con métodos compuestos (ver tabla 3).
Lee y Rendell usaron la teoría couple cluster en combinación con la reacción isodésmica HOOH + HOCl → HOOCl + H2O para calcular una ΔH◦ f,0K del HOOCl de 0,2±1,0 kcal mol−1 [22]. Empleando la misma reacción isodésmica, nosotros encontramos cifras de -0,2 (G4) y -0,1 (G4MP2) kcal mol−1. Nuestros resultados indican que la entalpía de formación para HOOCl es ligeramente exotérmica, un comportamiento similar a los resultados derivados por las energías de atomización a niveles de teoría CBS-QB3 y W1U (ver tablas 3 y 4).
Los nitro peróxidos HOONO y ClOONO fueron estudiados por McGrath y Rowland [27] al nivel teórico G2; nuestros resultados, estimados por esquemas de reacciones isodésmicas, son -2,7 y 31,4 kcal mol−1 para el HOONO y el ClOONO, respectivamente, al nivel teórico G4. Podemos destacar la alta precisión de los métodos compuestos al reportar los valores para la entalpía de formación de -2,6 y 31,6 para el HOONO y el ClOONO, respectivamente, con el otro método compuesto empleado (G4MP2). El valor para el ClOONO está en perfecto acuerdo con el reportado por McGrath y Rowland, mientras que el valor estimado para el HOONO es menor en aproximadamente 2,5 kcal mol−1.
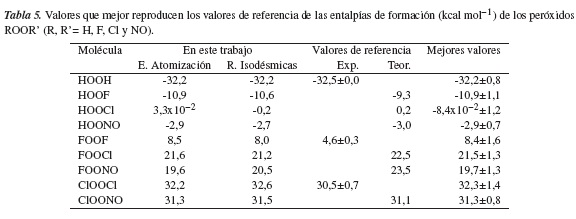
En la tabla 5 puede observarse la excelente convergencia entre los valores estimados para las entalpías de formación de los peróxidos ROOR' a partir de los esquemas de reacciones isodésmicas y las energías de atomización. Adicionalmente, cabe destacar que los métodos compuestos G4 y G4MP2 ofrecen valores muy cercanos a los valores promedios presentados en la tabla 3. Igualmente, en la Tabla 5 se presentan los valores calculados aquí que mejor reproducen los valores de referencia para las entalpías de formación de los peróxidos ROOR'. Como se ha señalado, el valor actual para el FOOF converge con el valor reportado por Jursic, de ΔH◦f,298K = 7,1 kcal mol−1 [49], que difiere notablemente de todos los demás valores teóricos reportados. En contraste, los valores de la entalpía de formación experimental y teórico para los compuestos HOOH, HOOF, HOOCl, HOONO, FOOCl, FOONO, ClOOCl y ClOONO han sido estimados con una elevada precisión en este trabajo.
4. Conclusiones
Se emplearon métodos compuestos ab initio de alto nivel para determinar con precisión las entalpías de formación a 298 K de los compuestos peróxidos ROOR' (R, R'= H, F, Cl y NO). Los valores estimados que mejor reproducen los valores de referencia para la ΔH◦f,298K de HOOH, HOOF, HOOCl, HOONO, FOOF, FOOCl, FOONO, ClOOCl y ClOONO son -32,2±0,8; -10,9±1,1; -8,4x10-2±1,2; -2,9±0,7; 8,4±1,6; 21,5±1,3; 19,7±1,3; 32,3±1,4 y -31,3±0,8 kcal mol−1, respectivamente. Las entalpías de formación obtenidas a partir de las energías de atomización y de los esquemas de reacción isodésmicas fueron comparadas con los valores reportados en la literatura, obteniendo una excelente concordancia para la mayoría de los compuestos estudiados. Las geometrías de equilibrio obtenidas al nivel de teoría B3LYP/6-31G(2df, p) corresponden a las estructuras caracterizadas experimentalmente y a las estructuras estudiadas por otros autores con niveles de teoría superiores.
Agradecimientos
C. B.-A. agradece el soporte económico recibido por la Vicerrectoría de Ciencia, Tecnología e Innovación (VCTI) de la Universidad Antonio Nariño, sede Bogotá, a través del proyecto No. 20131031.
Referencias
[1] I. N. Levine, Química Cuántica, Prentice Hall, 2001. [ Links ]
[2] E. Lewars, Computational chemistry: introduction to the theory and applications of molecular and quantum mechanics, Kuwer Academic Publishers, 2004. [ Links ]
[3] L. A. Curtiss, K. Raghavachari, P. C. Redfern, V. Rassolov, J. A. Pople, J. Chem. Phys. 1998, 109, 7764. [ Links ]
[4] L. A. Curtiss, P. C. Redfern, K. Raghavachari, J. Chem. Phys. 2007, 126, 084108. [ Links ]
[5] J. M. L. Martin, G. de Oliveira, J. Chem. Phys. 1999, 111, 1843. [ Links ]
[6] S. Parthiban, J. M. L. Martin, J. Chem. Phys. 2001, 114, 6014. [ Links ]
[7] T. H. Dunning Jr., J. Chem. Phys. 1989, 90, 1007. [ Links ]
[8] T. H. Dunning Jr., K. A. Peterson, D. E.Woon, "Correlation consistent basis sets for molecular calculations", in Encyclopedia of Computational Chemistry, P. von Ragué Schleyer (Ed.), Wiley & Sons, Chichester, UK, 1998. [ Links ]
[9] J. M. L. Martin, S. Parthiban; Chapter 2, pp. 31-65, In: Quantum Mechanical Prediction of Thermochemical Data, ed. J. Cioslowski and A. Szarecka; Understanding Chemical Reactivity Series, Vol. 22; Kluwer Academic Publishers, Dordrecht, The Netherlands, 2001; ISBN 07923-7077-5.99. [ Links ]
[10] D. W. Gunz, M. R. Hoffmann, Atmos. Environ. A Gen. Topics, 1990, 24, 1601. [ Links ]
[11] D. Gilbert, C. Colton (Eds.): Reactive Oxygen Species in Biological Systems, Kluwer Academic Press, New York, 2002. [ Links ]
[12] Z. Z. Rappoport (Ed.): The Chemistry of Peroxides, Volume 2, Parts 1, John Wiley & Sons, Chichester, England, 2006. [ Links ]
[13] G. Glockler, G. Matlack, J. Chem. Phys. 1946, 14, 504. [ Links ]
[14] J. R. Barker, S. W. Benson, D. M. Golden, Int.J. Chem. Kinet. 1977, 9, 31. [ Links ]
[15] M. W. Chase, C. A. Davies, J. R. Downey, D.J. Frurip, R. A. McDonald, A. N. Syverud, JANAF Thermodynamical Tables. J. Phys. Chem. Ref. Data, 1985, Supp. 1, 14. [ Links ]
[16] X. Luo, P. R. Fleming and T. R. Rizzo, J. Chem. Phys. 1992, 96, 5659. [ Links ]
[17] K. Young-Mi, L. Meehye, C. Wonil, L. Gangwoong, K. Kyung-Ryul, K. Shungo, Chemosphere, 2007, 69, 1638. [ Links ]
[18] G. S. Maciel, A. C. P. Bitencourt, M. Ragni, A. Vincenzo, J. Phys. Chem. A, 2007, 111, 12604. [ Links ]
[19] G. S. Maciel, A. C. P. Bitencourt, M. Ragni, V. Aquilanti, Int. J. Quantum Chem., 2007, 107, 2697. [ Links ]
[20] J. S. Francisco, J. Chem. Phys., 1993, 98, 2198. [ Links ]
[21] J. S. Francisco, J. Chem. Phys., 1993, 99, 10082. [ Links ]
[22] T. J. Lee, A. P. Rendell, J. Phys. Chem., 1993, 97, 6999. [ Links ]
[23] M. Weissman, L. G. S. Shum, S. P. Heneghan, S. W. Benson, J. Phys. Chem., 1981, 85, 2863. [ Links ]
[24] D. A. Dixon, D. Feller, C.-G. Zhan, J. S. Francisco, J. Phys. Chem. A, 2002, 106, 3191. [ Links ]
[25] D. Feller, D. A. Dixon, J. Phys. Chem. A, 2003, 107, 9641. [ Links ]
[26] D. Feller, K. A. Peterson, D. A. Dixon, J. Phys. Chem. A, 2008, 129, 204105. [ Links ]
[27] M. P. McGrath, F. S. Rowland, J. Phys. Chem., 1994, 98, 1061. [ Links ]
[28] M. J. Frisch, et al. Wallingford CT. Gaussian 09, Revision A.1, 2009. [ Links ]
[29] M. P. Badenes, C. J. Cobos, J. Mol. Struct. Theochem, 2008, 856, 59. [ Links ]
[30] L. A. Curtiss, P. C. Redfern, D. J. Frurip, Rev. Comput. Chem., 2000, 15, 147. [ Links ]
[31] A. D. McNaught and A. Wilkinson, IUPAC Compendium of Chemical Terminology, 2nd Edition, Blackwell Science, 1997. [ Links ]
[32] D. W. Rogers, Computational Chemistry Using the PC, John Wiley & Sons, 2003. [ Links ]
[33] A. G. Baboul, L. A. Curtiss, P. C. Redfern, K. Raghavachari, J. Chem. Phys., 1999, 110, 7650. [ Links ]
[34] L. A. Curtiss, P. C. Redfern, K. Raghavachari, J. Chem. Phys., 2007, 127, 124105. [ Links ]
[35] J. A. Montgomery, M. J. Frisch, J. W. Ochterski, G. A. Petersson, J. Chem. Phys., 1999, 110, 2822. [ Links ]
[36] G. Pelz, K. M. T. Yamada, G. Winnewisser, J. Mol. Spectrosc., 1993, 159, 507. [ Links ]
[37] R. H. Jackson, J. Chem. Soc. 1962, 4585. [ Links ]
[38] M. Birk, R. R.Friedl, E. A. Cohen, H. M. Pickett, S. P. Sander, J. Chem. Phys., 1989, 91, 6588. [ Links ]
[39] G. Schaftenaar, J.H. Noordik, J. Comput.-Aided Mol. Design, 2000, 14, 123. [ Links ]
[40] Y. D. Liu, R. G. Zhong, J. Mol. Struct. Theochem., 2006, 765, 143. [ Links ]
[41] C. Buendía-Atencio, J. Struct. Chem., 2012, 53, 582. [ Links ]
[42] C. Buendía-Atencio, Comput. Theor. Chem., 2011, 976, 161. [ Links ]
[43] C. Buendía-Atencio, C. J. Cobos, J. Fluorine Chem., 2011, 132, 474. [ Links ]
[44] O. Dorofeeva, V. S. Iorish, V. P. Novikov, D. B. Neumann, J. Phys. Chem. Ref. Data, 2003, 32, 879. [ Links ]
[45] A. J. Colussi, M. A. Grela, J. Phys. Chem., 1993, 97, 3775. [ Links ]
[46] M. A. Grela, A. J. Colussi, J. Phys. Chem., 1996, 100, 10150. [ Links ]
[47] P. Szakács, J. Csontos, S. Das, M. Kállay, J. Phys. Chem. A, 2011, 115, 3144. [ Links ]
[48] A. D. Kirshenbaum , A. V. Grosse , J. G. Aston, J. Am. Chem. Soc., 1959, 81, 6398. [ Links ]
[49] B. S. Jursic, J. Mol. Struct. Theochem., 1999, 459, 23. [ Links ]
[50] K. Andersson, B. O. Ross, Int. J. Quantum Chem., 1993, 45, 591. [ Links ]
[51] J. S. Francisco, J. Phys. Chem., 1994, 98, 5650. [ Links ]
[52] J. S. Francisco, J. Phys. Chem., 1996, 105, 3338. [ Links ]
[53] B. S. Jursic, J. Chem. Phys., 1997, 106, 2555. [ Links ]
[54] T. S. Dibble, J. S. Francisco, J. Am. Chem. Soc., 1997, 119, 2894. [ Links ]
[55] S. L. Nickolaisen, R. R. Friedl, S. P. Sander, J. Phys. Chem., 1994, 98, 155. [ Links ]
[56] S. P. Sander, J. Abbatt, J. R. Barker, J. B. Burkholder, R. R. Friedl, D. M. Golden, R. E. Huie, C. E. Kolb, M. J. Kurylo, G. K. Moortgat, V.L. Orkin, P. H. Wine, Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, Evaluation No. 17, JPL Publication 106, Jet Propulsion Laboratory, Pasadena, 2011, http://jpldataeval.jpl.nasa.gov (ref. 46). [ Links ]
[57] A. Karton, S. Parthiban, J. M. L. Martin, J. Phys. Chem A., 2009, 113, 4802. [ Links ]
[58] D. Feller, K. A. Peterson, D. A. Dixon, J. Phys. Chem. A, 2010, 114, 613. [ Links ]
