










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkCiencia en Desarrollo
versão impressa ISSN 0121-7488
Ciencia en Desarrollo vol.7 no.1 Tunja jan./jun. 2016
Efecto de la concentración de Ga sobre las propiedades electrónicas del CuIn1-XGaXSe2
Ga Concentration Effect on the CuIn1-xGaxSe2 Electronic Properties
D. A. Rasero Causila,*
A. A. Portacio Lamadridb
J. A. Rodríguezc
a Departamento de Ciencias Naturales, Universidad Surcolombiana, Neiva, Colombia.
* Autor de correspondencia: diegorasero@gmail.com.
b Departamento de Matemáticas y Física, Universidad de los Llanos, Villavicencio, Colombia.
c Departamento de Física, Universidad Nacional de Colombia, Bogotá, Colombia.
Recepción: 04-sep-2015 Aceptación: 20-ene-2016
Resumen
Se reportan cálculos de propiedades electrónicas del compuesto CuIn1-xGaxSe2 (x = 0,0; 0,2; 0,4; 0,6; 0,8; 1,0), usando el método Tight-Binding (TB) y Virtual Crystal Approximation (VCA). Se considera el caso ideal y con las distorsiones tetragonal (η) y aniónica (μ). En ambos casos, el CuIn1-xGaxSe2 es un semiconductor directo en Γ, para todas las concentraciones. Se encontró que el Crystal Field Splitting (CFS) en el punto Γ depende principalmente de la distorsión tetragonal. El CFS es positivo para x < 0,32 y negativo para x > 0,32. Este comportamiento se debe a que cuando aumenta x, la celda unitaria se contrae, acercando el pseudoátomo (In,Ga) al átomo de Se.
Palabras clave: CuIn1-xGaxSe2, Tight-Binding, aproximación de cristal virtual, Crystal Field Splitting.
Abstract
This paper reports some calculations of the electronic properties of CuIn1-xGaxSe2 (x = 0.0, 0.2, 0.4, 0.6, 0.8, 1.0) compound, by using the Tight-Binding (TB) method and Virtual Crystal Approximation (VCA). It is considered the ideal case and with the tetragonal (η) and anionic (μ) distortions. In both cases, the CuIn1-xGaxSe2 is a direct semiconductor at Γ, for all concentrations.It was found that the Crystal Field Splitting (CFS) at the Γ point depends mainly on the tetragonal distortion. The CFS is positive for x < 0,32 and negative for x > 0.32. This behavior is due that when x is increasing, the unit cell shrinks, approaching the pseudo-atom (In,Ga) to the Se atom.
Key words: CuIn1-xGaxSe2, Tight-Binding, Virtual Crystal Approximation, Crystal Field Splitting.
1. Introducción
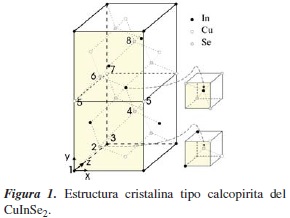
Entre las diferentes fuentes de energía, la solar es una buena elección frente a los combustibles fósiles, ya que no contamina el ambiente y se puede utilizar durante gran parte del año. Para aprovechar esta o cualquier otra fuente de energía se necesita del generador adecuado. Los generadores fotovoltáicos son una alternativa de producción de energía eléctrica; con estos se puede complementar la energía generada, por ejemplo, por hidroeléctricas, que están siendo limitadas por la influencia del clima, de las lluvias, y últimamente afectadas por fenómenos de cambio climático; también se puede complementar la energía de termoeléctricas, que consumen combustible fósil, agotando las reservas y, además, contaminan el medioambiente. Con el compuesto ternario CuInSe2 (CIS) en estructura calcopirita (ver figura 1) se han construido celdas solares fotovoltáicas de bajo costo y eficiencias cercanas al 16% [1]. Aleando los semiconduntores CuInSe2(de gap directo 1,04 eV) y CuGaSe2 (de gap directo 1.70 eV) en estructura policristalina se pueden construir celdas con eficiencias cercanas al 19% [2-7]; esta aleación, conocida como CuIn1-xGaxSe2 (CIGS), conforma la capa absorbente de la celda.
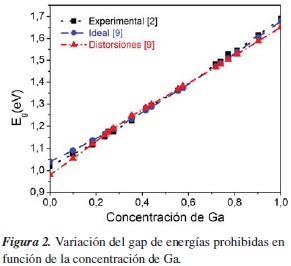
Los resultados experimentales [8] muestran que el gap de energías prohibidas Eg de esta aleación varía cuadráticamente con la concentración x de Ga. El parámetro de curvatura (bowing parameter) experimental es bexp = 0,107 eV. En un reporte previo [9] hemos calculado los parámetros Tight-Binding (TB) que reproducen esta dependencia cuadrática y la estructura electrónica para la concentración x = 0,6.
En este trabajo calculamos la estructura electrónica y la variación del Crystal Field Splitting (CFS) principal del CuIn1-xGaxSe2 para las concentraciones de Ga, x = 0,0; 0,2; 0,4; 0,6; 0,8 y 1,0; usando el método TB y Virtual Crystal Approximation (VCA).
2. Aspectos teóricos
2.1. Estructura cristalina
La celda unitaria del CuInSe2, CuIn1-xGaxSe2 y CuGaSe2 es tetragonal centrada en el cuerpo, con ocho átomos, como se muestra en la figura 1. En el caso ideal cada catión (anión) está en el centro de un tetraedro determinado por sus vecinos cercanos y c = 2a. En un caso más real se pueden considerar dos distorsiones: la distorsión aniónica μ, que implica que los aniones no están exactamente en el centro de dichos tetraedros, y la distorsión tetragonal η = c/2a, que impílica c ≠ 2a. Según esto, en el caso ideal μ = 0,25 y η = 1.
Para CuInSe2 μ = 0,224 y η = 1,004, para CuGaSe2 μ = 0,248 y η = 0,982.
2.2. Método Tight-Binding
El hamiltoniano se construye bajo el modelo TB de Slater and Koster [10], con la modificación de Blom et al. [11] y de Rodríguez [12, 13], en el cual se usa la base de funciones de Bloch:
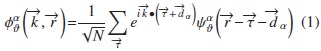
En esta expresión,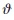 identifica los orbitales atómicos α, los átomos de la celda, y
identifica los orbitales atómicos α, los átomos de la celda, y 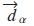 representa la posición del átomo
representa la posición del átomo 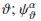 son orbitales atómicos localizados y
son orbitales atómicos localizados y 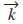 es el vector de onda. Se toman orbitales s y p para In, S y Se, y orbitales s, p y d para Cu. No se considera la interacción spin-órbita. Las amplitudes de transición del electrón de un órbital a otro están dadas por
es el vector de onda. Se toman orbitales s y p para In, S y Se, y orbitales s, p y d para Cu. No se considera la interacción spin-órbita. Las amplitudes de transición del electrón de un órbital a otro están dadas por
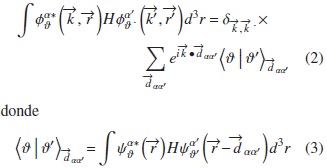
La suma que aparece en (2) involucra únicamente interacciones hasta primeros vecinos. La matriz hamiltoniana que resulta tiene dimensión 42 × 42 y se diagonaliza numéricamente, a lo largo de algunas direcciones de alta simetría de la primera zona de Brillouin (PZB).
2.3. Virtual Crystal Approximation
En esta aproximación se considera la aleación como una estructura periódica ordenada. Se adopta la descripción de Hill [14], en la cual el desorden de corto alcance no juega un papel importante en la variación del gap Eg con la concentración x. Bajo esta aproximación se considera que en CuIn1-xGaxSe2 existe un pseudo-átomo (In,Ga). Para x = 0,0 se tiene CuInSe2, y para x = 1,0, CuGaSe2. Se propone, según Olgin [15], que el parámetro s del (In,Ga) varíe de la siguiente manera:
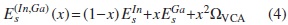
Donde 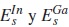 son los parámetros on-sites del In y Ga en el CuInSe2 y en el CuGaSe2, respectivamente; ΩVCA es el parámetro VCA de curvatura, introducido para reproducir el comportamiento cuadrático de la curva experimental Eg(x), propuesto de la siguiente forma:
son los parámetros on-sites del In y Ga en el CuInSe2 y en el CuGaSe2, respectivamente; ΩVCA es el parámetro VCA de curvatura, introducido para reproducir el comportamiento cuadrático de la curva experimental Eg(x), propuesto de la siguiente forma:
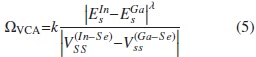
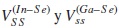 son los parámetros de interacción entre orbitales s, a primeros vecinos, de In y Se en el CuInSe2, y de Ga y Se en el CuGaSe2, respectivamente. Se propone que los parámetros p del (In,Ga) varíen linealmente con x, esto es:
son los parámetros de interacción entre orbitales s, a primeros vecinos, de In y Se en el CuInSe2, y de Ga y Se en el CuGaSe2, respectivamente. Se propone que los parámetros p del (In,Ga) varíen linealmente con x, esto es:
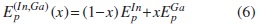
Para determinar ΩVCA se realiza un proceso de minimización de la función:
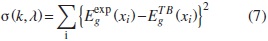
Donde 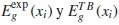 son los valores del gap, experimental y teórico (usando la aproximación TB y VCA) para la concentración xi respectivamente.
son los valores del gap, experimental y teórico (usando la aproximación TB y VCA) para la concentración xi respectivamente.
3. Resultados y Discusión
3.1. Gap de energías prohibidas
En la figura 2 se muestra la variación de Eg como una función de la concentración de Ga. Los rectángulos indican el resultado experimental [2]; con círculos y triángulos se indican los resultados teóricos, calculados en un trabajo previo con el método Tight-Binding [9], para el caso ideal y con distorsiones, respectivamente.
Eg varía de manera cuadrática con x [9]. Los datos experimentales indican que
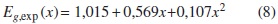
Los parámetros VCA de curvatura para el caso ideal y con distorsiones obtenidos según nuestros cálculos son ΩVCA, ideal = -0,29204 eV y ΩVCA, dist = -0,13219 eV, con los que se obtiene
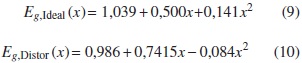
Como se observa en la figura 2, nuestros resultados concuerdan bastante bien con los datos experimentales.
3.2. Estructura de bandas de energía
Los cálculos de estructura electrónica se muestran en la figura 3, en la que se comparan las bandas ideales (línea continua) y con distorsiones (línea discontinua) para las concentraciones de Ga, x = 0,0; 0,2; 0,4; 0,6; 0,8 y 1,0.
El cero de la escala de energías se ha colocado en el máximo de la banda de valencia (BV). Las primeras 26 regiones energéticas corresponden a la BV, y las 16 restantes, a la banda de conducción (BC).
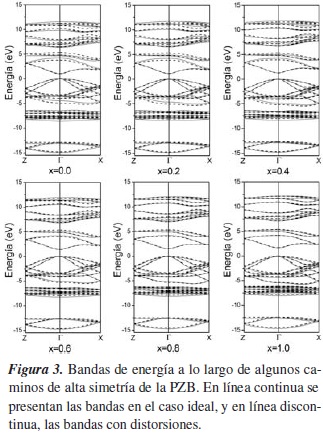
Nuestros resultados indican que tanto en el caso ideal como con distorsiones, el CuIn1-xGaxSe2 es un semiconductor directo en Γ, para todas las concentraciones. Este resultado concuerda con el de Rodríguez, para x = 0,0y x = 1,0, [12,13]
En ambos casos, el borde inferior de la BC sube, aumentando el valor del gap. La forma de la parte superior de la BV y de la inferior de la BC, cerca del punto Γ, es parabólica, y para cada concentración no varía de un caso a otro, indicando que la masa efectiva de los portadores de carga no cambia.
3.3. Crystal field Spliting principal en Γ
En los semiconductores tipo zinc-blenda (ZB) el borde superior de la BV es un triplete (debido a los orbitales p-anión, que predominan en esta zona). Las calcopiritas presentan dos cationes en nuestro caso, el Cu y el pseudo-átomo (In,Ga); por tal razón, el triplete se desdobla en un single-te y un doblete. La diferencia de energía entre estos, ΔCFS = Edoblete - Esinglete, se llama Crystal Field Splitting principal (CFS). El valor del CFS se debe físicamente a tres causas [16,17]: la existencia de dos cationes, la distorsión tetragonal η y la distorsión aniónica μ.
En la figura 4 se muestra la variación del CFS como una función de x.
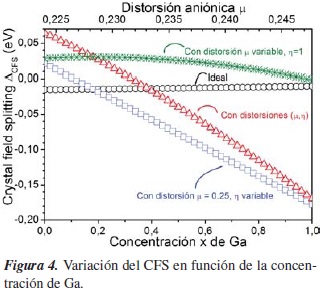
Para el caso ideal (círculos), el CFS se mantiene aproximadamente constante y aparece por la existencia de los cationes Cu e (In,Ga). Además, siempre es negativo, indicando que el singlete está por encima del doblete.
La pequeña variación del CFS se debe al cambio que experimenta la constante de red y los parámetros TB del (In,Ga) a medida que la concentración cambia. Cuando se consideran las distorsiones (triángulos), el CFS varía cuadráticamente con x, de acuerdo con el ajuste (en eV):
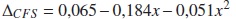
Esta variación se debe principalmente a los cambios que sufren las distorsiones η y μ cuando x varía. Para la concentración x = 0,32 (μ = 0,232, η = 0,997), ΔCFS = 0, indicando que el borde superior de la BV es un triplete. Para x < 0,32, el CFS es positivo, mientras que para x > 0,32 el CFS es negativo. El cambio de posición del doblete y el singlete se muestran en la figura 5.

En la figura 4 también se muestra como cambia el CFS cuando μ (en asterísticos) y η (en rectángulos) varían de manera independiente. Esto indica que la distorsión tetragonal es la principal responsable de la variación del CFS.
En términos de la posición de los átomos en la celda unitaria (ver figura 1), el efecto de la distorsión η consiste en disminuir la longitud de enla ceSe (In,Ga). Así, a medida que la concentración de Ga aumenta, la interacción entre estos átomos también aumenta, dando origen al cambio de signo del CFS.
4. Conclusiones
El estudio del compuesto CuIn1-xGaxSe2 para las concentraciones x = 0,0-1,0, usando TB y VCA, muestra que este material es semiconductor directo en Γ. El CFS varía cuadráticamente con x. Para el caso ideal, el CFS siempre es negativo, y para el caso con distorsiones cambia de signo, lo cual se debe a la interacción de los átomos de Se con el pseudo-átomo (In,Ga).
Agradecimientos
A la Universidad Nacional de Colombia, por su apoyo financiero durante el desarrollo de este trabajo.
Referencias
[1] J. Hedstrom, H. Ohlsen, M. Bodegard, A. Kylner, L. Stolt, D. Hariskos, M. Ruckh, and W. Schock, "ZnO/CdS/Cu(In,Ga)Se2 thin film solar cells with improved performance", in Proceedings of the 23rd IEEE Photovoltaic Specialists Conference, pp. 364-371, May 1993. [ Links ]
[2] M. Contreras, B. Egaas, K. Ramanathan, J. Hiltner, A. Swartzlander, F. Hasoon, and R. Noufi, "Progress toward 20% efficiency in Cu(In,Ga)Se2 polycrystalline thin-film solar cells", Prog. Photovolt: Res. Appl., vol. 7, pp. 311-316, August 1999. [ Links ]
[3] M. Contreras, A. M. Gabor, A. L. Tenant, S. Asher, J. Tuttle, and R. Noufi, "Accelerated publication 16.4% total-area conversion efficiency thin-film polycrystalline MgF2/ZnO/CdS/Cu(In,Ga)Se2/Mo solar cell", Prog. Photovolt: Res. Appl., vol. 2, pp. 287-292, October 1994. [ Links ]
[4] J. R. Tuttle, M. A. Contreras, A. M. Gabor, K. R. Ramanathan, A. L.Tennant, D. S. Albin, J. Keane, and R. Noufi, "Perspective on High-efficiency Cu(In, Ga)Se2-based Thin-film Solar Cells Fabricated by Simple, Scalable Processes", Prog. Photovolt: Res. Appl., vol. 3, 383-391, November 1995. [ Links ]
[5] J. R. Tuttle, J. S. Ward, A. Duda, T. A. Berens, M. A. Contreras, K. R. Ramanathan, A. L. Tennant, J. Keane, E. D. Cole, K. Emery, and R. Nouri, "The Performance of Cu(In,Ga)Se2-Based Solar Cells in Conventional and Concentrator Applications", Mater. Res. Soc. Symp., vol. 426, 143-151, 1996. [ Links ]
[6] J. R. Tuttle, M. Contreras, A. Tennant, D. Albin, and R. Noufi, "High efficiency thin-film Cu (In, Ga) Se2-based photovoltaic devices: progress towards a universal approach to absorber fabrication", in Proceedings of the 23rd IEEE Photovoltaic Specialists Conference, pp. 415-421, May 1993. [ Links ]
[7] K. R. Ramanathan, M. A. Contreras, C. L. Perkins, S. Asher, F. S. Hasoon, J. Keane, D. Young, M. Romero, W. Metzger, R. Noufi, J. S. Ward, and A. Duda, "Properties of 19.2% efficiency ZnO/CdS/CuInGaSe2 thin-film solar cells", Prog. Photovolt: Res. Appl., vol.11, pp. 225-230, June 2003. [ Links ]
[8] R. W. Birkmire, J. R. Sites, "Polycrystalline compound thin film devices: Laboratory cells to modules", in: IEEE (Ed.), 28th IEEE PVSC (Photovoltaic Specialist Conference), IEEE, Anchorage, AK, USA, 2-11, 2000. [ Links ]
[9] T. Suárez, D. Rasero, R. Jiménez, and J. Arbey Rodríguez, "Cu(InGa)Se2: Un estudio de la estructura electrónica usando Tight-Binding, Aproximación de Cristal Virtual y Método de Montecarlo", Rev. Col. Fis., vol. 41, pp. 268- 270, abril 2009. [ Links ]
[10] J. C. Slater, and G. F. Koster, "Simplified LCAO Method for the Periodic Potential Problem", Phys. Rev., vol. 94, pp. 1498-1524, June 1954. [ Links ]
[11] F. A. P. Blom,"Determination of the Fermi Surface in CdSnAs using a Tight-Binding Model for Chalcopyrites", Phys. Rev. B, vol. 32, pp. 2334-2336, August 1985. [ Links ]
[12] J. A. Rodríguez, "Estructura de Bandas de las Calcopiritas basadas en Cobre", Tesis doctoral, Departamento de Física, Universidad Nacional de Colombia, Ciudad Universitaria, Bogotá, D. C., 1999. [ Links ]
[13] J. A. Rodríguez, L. Quiroga, A. Camacho, and R. Baquero, "Electronic Band Structure of CuInSe2: Bulk and (112) surface", Phys. Rev. B,, vol. 59, pp. 1555-1558, January 1999. [ Links ]
[14] R. Hill, "Energy-gap Variations in Semiconductor Alloys", J. Phys. C: Solid State Phys., vol. 7, pp. 521-526, February 1974. [ Links ]
[15] R. D. Olguín, "Estructura Electrónica de Compuestos Semiconductores Nuevos II-VI: Superficies y Heteroestructuras", Tesis doctoral, Departamento de Física, Centro de Investigaciones y de Estudios Avanzados del IPN (CINVESTAV), México, D. F., México, 1996. [ Links ]
[16] J. Jaffe, A. Zunger, "Electronic Structure of the Ternary Chalcopyrite Semiconductors CuAlS2, CuGaS2, CuInS2, CuAlSe2, CuGaSe2, and CuInSe2", Phys. Rev. B, vol. 28, pp. 5822-5847, November 1983. [ Links ]
[17] J. Jaffe, and A. Zunger, "Theory of the Bandgap Anomaly in ABC2 Chalcopyrite Semiconductors", Phys. Rev. B, vol. 29, pp. 1882-1906, February 1984. [ Links ]

