











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink1 Introducción
La obtención de imágenes in vivo se ha convertido en un área de mucha importancia en el campo de la medicina [1]. En la actualidad, aproximadamente el 30 % de los pacientes que son sometidos a procedimientos de adquisición de imágenes por resonancia magnética nuclear (RMI) u otras técnicas, son inyectados con medicamentos para contrastar sectores del cuerpo cuyas imágenes no son claras, acentuando la importancia y preferencia por el uso de técnicas teragnósticas [2].
Los agentes de contraste, que en su estructura química tienen iones lantánidos, son los más usados en la mayoría de las técnicas de imagenología clínica. Desde la salida del Magnevist® (Gd-DTPA dimeglumina) en 1988, un agente de contraste usado para RMI, se han encontrado diferentes efectos secundarios por su uso, entre los cuales se destaca la fibrosis sistémica nefrogénica (FSN), una enfermedad relacionada con la acumulación de gadolinio en pacientes con deficiencias renales y que fue detectada por primera vez en el 2006 [3]; adicionalmente, uno de los principales efectos del uso de agentes de contraste ha sido la retención de metales pesados en el sistema nervioso central y óseo [3]. Así, este tipo de complicaciones reduce aún más el número de pacientes viables para el suministro de este tipo de compuestos con el fin de esclarecer un determinado diagnóstico. Actualmente se siguen usando, mayoritariamente, los mismos compuestos que fueron descubiertos entre 1988 y 1995, con tan sólo algunos pocos aprobados para uso clínico [3]. Por lo cual, los efectos secundarios siguen presentes y son considerados como efectos colaterales que los pacientes actualmente deben aceptar como contraindicaciones. Dentro de los complejos lantánidos más usados se encuentran el ácido gadotérico (GdDOTA) y el ácido gadopentético (GdDTPA), ligandos multidentados macrocíclico y lineal respectivamente, de altos costos de producción debido a sus bajos rendimientos de obtención [1]. Las estabilidades que proveen los ligantes polidentados vienen de la mano del carácter oxófilo de los metales lantánidos, de tal manera que siempre se han empleado grupos carboxilatos, fosfatos y éteres de corona modificados como puntos de coordinación fundamentales [2].
Por tal motivo, el desarrollo de nuevos agentes de contraste con iones lantánidos (III) sigue siendo un área de investigación aún muy activa [4], en donde la obtención de moléculas con mayores relajatividades molares, mayor estabilidad termodinámica-cinética y mayores rendimientos cuánticos, son necesarios para dar solución a los principales retos a bordar, como las altas dosis, la acumulación de metales en el sistema óseo y las afectaciones renales provocadas en pacientes con cuadros clínicos complejos [1].
Los ligandos flexibles han mostrado ser promisorios para el mejoramiento de los parámetros modulables de diferentes agentes de contraste [5]. El ácido gadopentético y gadotérico presentan ligando relativamente flexibles (ver figura 1), sin embargo, la mayor adaptabilidad al radio iónico de los metales en diferentes condiciones fisiológicas puede incrementar la estabilidad termodinámica y cinética, disminuyendo los efectos adversos de los actuales agentes [6].
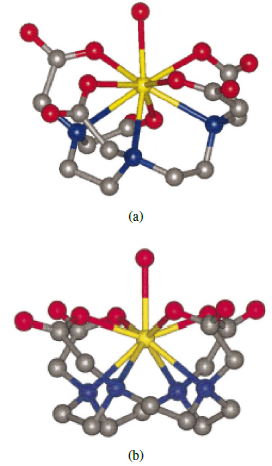
Figura 1 Estructura cristalina de (a). Ácido gadopentético y (b). Ácido gadotérico (esferas amarillas = Gd; esferas grises = C; esferas azules = N; esferas rojas = O) [5].
Adicionalmente, los agentes de contraste multimodales (compuestos útiles para diferentes técnicas de imagenología en simultáneo) son bastante llamativos, ya que se pueden generar múltiples imágenes con una sola dosis suministrada, disminuyendo el uso de más agentes y complementando el diagnóstico clínico [6].
En consecuencia, en el presente trabajo de investigación, se presenta la síntesis de nuevos complejos lantánidos con ligandos polidentados flexibles, la evaluación de su potencial aplicación en el diagnóstico clínico como agentes de contraste moleculares para RMI y la evaluación de su actividad biológica contra cepas bacterianas, para determinar su uso como agentes de contraste óptico celular.
2 Materiales y métodos
2.1 Reactivos
Los reactivos empleados para las síntesis y los ensayos fueron adquiridos de las casas comerciales Sigma Aldrich, Alfa Aesar y Merck Millipore, no se realizó ningún tipo de purificación previa a su uso. Todos los solventes fueron usados en grado analítico. El agente Dotarem® fue adquirido del proveedor RX S.A como fármaco comercial para radiología.
2.2 Equipos
Los espectros infrarrojos fueron tomados entre 4000-600 cm-1 en un equipo Shimadzu Affinity-1 (FT-IR) mediante reflectancia totalmente atenuada (ATR), para la zona inorgánica entre los 600-250 cm-1 se usó un espectrofotómetro Thermo Scientific NICOLET 6700 (FT-IR) mediante la técnica de pastilla de KBr. La determinación del porcentaje de iones lantánidos se realizó mediante titulación colorimétrica con naranja de xilenol, empleando un espectrofotómetro UV-visible Evolution 220 Thermo Scientific con accesorio Peltier de mono celda para el control de la temperatura [7]. Los puntos de fusión fueron determinados usando un equipo automático OptiMelt MPA100. Las conductividades molares de los complejos lantánidos fueron determinadas con el uso de un conductímetro OrionTM 131S. Los rendimientos cuánticos de fluorescencia de los compuestos fueron determinados con el uso de un espectrofluorímetro JASCO FP-8500 utilizando fluoresceína como patrón. Los espectros 1H RMN y 13C RMN fueron tomados en Cloroformo-d1 y DMSO-d6 a 25 °C en un espectrofotómetro Bruker Ultrashield Avance II 400 operando a 400 MHz, los desplazamientos químicos (5) son reportados en unidades de ppm, respecto al patrón TMS. La completa asignación de todas las señales RMN de los nuevos compuestos se apoyó en el análisis de los espectros bidimensionales obtenidos a través de las técnicas HMBC y HSQC; el mismo equipo fue empleado para la determinación de las relajatividades molares de los complejos obtenidos, usando la secuencia "recuperación de saturación" con una intensidad de campo de 9 T a 300 K. Los análisis térmicos se llevaron a cabo en un dispositivo TGA 550 TA Instruments en atmósfera de nitrógeno con rampa de temperatura de 5 °C/min. Los espectros de masas fueron tomados en un equipo Shimadzu-GCMS-QP2010 con ionizador por impacto electrónico a 70 eV.
2.3 Síntesis de ligandos
Se sintetizaron dos ligandos polidentados lineales para evaluar el efecto de la flexibilidad, el número de coordinación y presencia de fluoróforo en los parámetros importantes de contraste.
2.3.1 Síntesis del ligando dietil 2,2'-(butano-1,4-dilbis((2-hidroxibencil)azanedil))diacetato
La ruta sintética del ligando L se muestra en la figura 2.
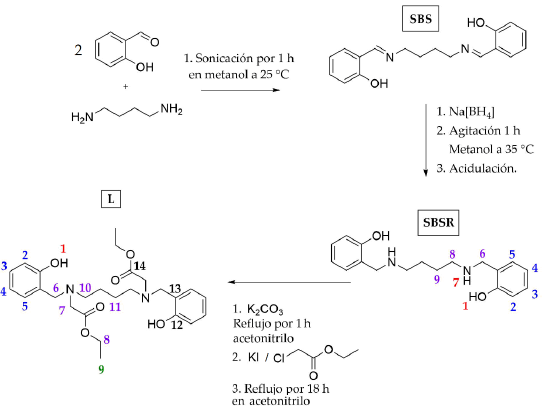
Se realizó una mezcla de 2 mL de salicilaldehído (19.2 mmol) y 960 µL de 1,4-diaminobutano (9.6 mmol) en un erlenmeyer que contenía 2 mL de metanol. La solución resultante se llevó a sonicación por 1 hora a 25 °C; después de este tiempo, el sistema fue enfriado a 4 °C por 30 minutos y el sólido fue filtrado y lavado, con 100 mL de una mezcla fría 1:1 metanol:agua y con 50 mL de metanol frío. El compuesto fue identificado como N,N'-bis(saliciliden)-1, 4-butanodiamina (SBS) con un punto de fusión de 88 °C - 89 °C próximo al reportado en la literatura [8], con un rendimiento del 90 %.
Posteriormente, 2490 g (8.4 mmol) de SBS fueron sometidos a reducción con borohidruro de sodio 0.673 g (17.8 mmol) en metanol. Se observó efervescencia inmediata al agregarse el borohidruro de sodio a la solución metanólica de SBS; la mezcla se agitó a 35 °C por 1 hora, tiempo en el que la solución dejó de efervescer y se tornó incolora. Se agregó agua fría al sistema para promover la precipitación del compuesto reducido SBSR, el sistema fue enfriado a 4 °C por 30 minutos y el sólido fue filtrado y lavado con 100 mL de una mezcla fría 1:1 metanol:agua y con 50 mL de metanol frío. El compuesto fue identificado a través del punto de fusión y los espectros RMN protónico e infrarrojo en comparación con reportes previos [8]. Polvo blanco (rendimiento: 70 %). p.f.: 118-119 °C. IR (ATR cm-1): и 3286 d, 2931 d, 2902 d, 2850 d, 2813 m, 1597 m, 1452 m, 1395 m, 1380 m, 1337 m, 1272 m, 1252 m, 1238 m, 1192 m, 1163 m, 1113 m, 1091 m, 1040 m, 997 m, 975 d, 939 , 866 m, 839 m, 751 f, 721 m. 1H-RMN (CDCl3, 400 MHz): 5 (ppm) 1.63 (q, J = 3.3 Hz, 4H, H9), 2.72 (t, J = 6.0Hz, 4H, H8), 4.02 (s, 4H, H6), 6.81 (td, J = 7.4 Hz, 1.2 Hz, 2H, H4), 6.86 (dd, J = 8.2 Hz, 1.1 Hz, 2H, H2), 7.01 (dd, J = 7.4 Hz, 1.6 Hz, 2H, H5), 7.2 (td, J = 7.8 Hz, 1.7 Hz, 2H, H3).
Se dispuso en un balón de dos bocas 1.502 g (5.0 mmol) de SBSR, 10.1 g (73.1 mmol) de carbonato de potasio seco y 50 mL de acetonitrilo seco. El sistema se dejó a reflujo por 1 hora y posteriormente se agregaron 0.830 g (5.0 mmol) de yoduro de potasio seco junto con 1.11 mL (10.4 mmol) de cloroacetato de etilo. El sistema continuó en reflujo por otras 18 horas. Una vez terminado el tiempo de reacción, se filtraron los sólidos inorgánicos insolubles y el solvente fue removido con evaporación a presión reducida. El sólido obtenido del crudo de reacción tuvo un color amarillo pálido y fue recristalizado en metanol. Cristales incoloros (rendimiento: 65 %). C26H36N2O6. p.f.: 250-251 °C. IR (pastilla KBr cm-1): и 3285 d, 3050 m, 2982 m, 2954 m, 2926 m, 2909 m, 2858 m, 1738 f, 1615 m, 1587 f, 1509 m, 1492 f, 1458 m, 1424 m, 1374 m, 1313 m, 1274 m, 1246 m, 1190 m, 1122 m, 1094 m, 1033 f, 987 f, 948 f, 936 m, 892 m, 869 m, 853 f, 808 d, 774 m, 752 f, 713 m, 662 d, 629 d, 589 d, 500 m, 450 f, 400 m, 320 f, 300 m, 281 f. 1H-RMN (CDCl3, 400 MHz): 5 (ppm) 1.27 (t, J = 7.1 Hz, 6H, H9), 1.49 (q ancho, 4H, H11), 2.53 (t ancho, 4H, H10), 3.31 (s, 4H, H6), 3.79 (s, 4H, H7), 4.18 (c, J = 7.1 Hz, 4H, H8), 6.76 (td, J = 7.4 Hz, 0.7 Hz, 2H, H4), 6,83 (d, J = 8.0 Hz, 2H, H2), 6.95 (d, J = 7.1 Hz, 2H, H5), 7.17 (td, J = 7.7 Hz, 0.8 Hz, 2H, H3), 10.02 (s, 2H, H1). 13C-RMN (CDCl3, 100 MHz): 5 14.3 (C9), 24.3 (C11), 53.2 (C10), 54.5 (C6), 58.2 (C7), 61.2 (C8), 116.4 (C2), 119.3 (C4), 121.8 (C13), 129.1 (C3), 129.2 (C5), 157.7 (C12) 170.8 (C14). EM (IE, 70 eV): m/z472 [M+].
2.3.2 Síntesis del ligando 4,4'-((1E,1'E)-hidracin- 1.2- dilidenbis(metanoiliden))bis(bencen-1.3- diol)
El segundo ligando, se sintetizó como fluoróforo F, cuya síntesis se resume en la figura 3.
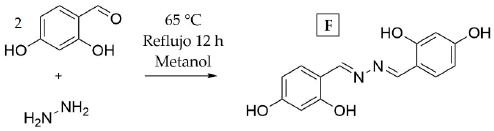
0.525 g (3.6 mmol) de 2,4-dihidroxibenzaldehído y 0.20 mL (4.1 mmol) de hidrazina monohidratada se llevaron a reflujo en 20 mL de metanol por 12 horas. Una vez finalizada la reacción, confirmada por cromatografía de placa delgada, se agregaron 50 mL de agua fría para inducir precipitación de la azina del 2,4-dihidroxibenzaldehído (F) de color amarillo; el sólido fue lavado con 20 mL de una mezcla fría 1:1 metanol:agua. El compuesto se identificó a través de espectrometría de masas, en donde se observó el ion molecular a m/z 272 [M+] con fragmentos característicos (ver material suplementario). El rendimiento obtenido fue del 50 %. C14H12N2O4. IR (ATR cm-1): v 3498 d, 3220 d, 1612 f, 1580 m, 1508 m, 1445 m, 1310 m, 1248 m, 1212 m, 1176 m, 1118 m, 985 m, 850 m, 781 m, 745 m, 682 m, 646 m, 568 m. Los resultados de la caracterización son acordes a lo reportado en investigaciones previas [9, 10].
2.4 Síntesis de complejos lantánidos
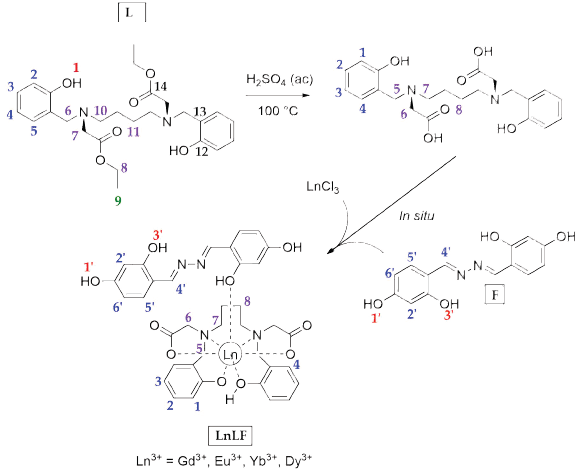
Cuatro complejos lantánidos de con iones Gd(III), Eu(III), Dy(III) e Yb(III) fueron sintetizados a partir de los ligandos obtenidos. La ruta sintética para la obtención de los cuatro complejos se muestra en la figura 4.
Se disolvieron 0.378 g (0.8 mmol) de L en 100 mL de ácido sulfúrico 0.5 mol.L-1 (50 mmol) y se dejó a reflujo a 100 °C por 2 horas. Posteriormente, se dejó enfriar el sistema a 50 °C ajustando el pH a 7.0 con hidróxido de sodio 1.0 mol.L-1. A esta solución se adicionó lentamente el respectivo tricloruro de lantánido (0.297 g GdCb; 0.293 g EuCb; 0.302 g DyCl3 y 0.310 g YbCb) (0.8 mmol). Finalmente, se agregaron 10 mL de una solución metanólica 0.08 mol.L-1 de F (0,8 mmol) agitando el sistema por 3 horas. En todos los casos, después de concentrar la solución hasta 10 mL aproximadamente, se observó un precipitado el cuál fue filtrado. El sólido obtenido fue lavado con una solución hexano:metanol 60:40 frío y secado al aire libre.
1; polvo amarillo claro (rendimiento: 25 %). C37H37N4O10Gd, porcentaje de gadolinio Gd 17.65 ± 0.8 (calc. 18.7) %. p.f. > 300 °C. IR (ATR cm-1): v 3518 d, 3465 d, 3207 d, 2984 d, 2932 d, 2890 d, 1725 f, 1611 f, 1586 m, 1509 m, 1446 f, 1328 m, 1308 m, 1254 m, 1214 m, 1174 m, 1121 m, 974 m, 843 m, 775 m, 684 m, 642 m. 1H-RMN (DMSO-d6, 400 MHz): δ 1.54 (s, ancho, H8), 2.52 (s, ancho, H7), 2.94 (s, ancho, H6), 6.16 (s, ancho, 2H, H6'), 6.25 (s, ancho, 2H, H2'), 6.74 (m, ancho, H1 y H3), 7.13 (m, ancho, H4), 7,22 (d, J = 6.8 Hz, 2H, H5'), 7.37 (m, ancho, H2), 8.53 (s, ancho, 2H, H4'), 9.59 (s, ancho, H1'). Λ (DMSO, 28 °C) (Ω-1.cm2.mol-1): 59.6.
2; polvo amarillo claro (rendimiento: 30 %). C37H37N4O10Eu, porcentaje de europio Eu 19.18 ± 0.8 (calc. 18.1) %. p.f. > 300 °C. IR (ATR cm-1): v 3478 d, 3203 d, 3030 d, 2939 d, 2017 d, 1917 m, 1896 m, 1754 f, 1722 f, 1695 m, 1624 m, 1588 m, 1455 m, 1393m, 1317m, 1276m, 1250m, 1194 m, 1122 m, 979 m, 892 m, 845 m, 764 m, 687 d, 651 d, 651d, 543 d. 1H-RMN (DMSO-d6, 400 MHz): δ 1.73 (s, ancho, 4H, H8), 3.13 (s, ancho, 4H, H7), 3.91 (s, ancho, 4H, H6), 6.37 (m, ancho, H2' y H6'), 6.89 (t, J = 7.18 Hz, 2H, H1), 6.93 (d, J = 7.9 Hz, 2H, H3), 7.32 (m, ancho, 4H, H2 y H4), 7,51 (m, ancho, H5'), 8.78 (d, J = 7.1 Hz, 2H, H4'), 9.77 (s, ancho, H1')A (DMSO, 28 °C) (Ω-1.cm2.mol-1): 65.1.
3; polvo amarillo claro (rendimiento: 37 %). C37H37N4O10Dy, porcentaje de disprosio Dy 19.48 ± 0.6 (calc. 19.1) %. p.f. > 300 °C. IR (ATR cm-1): v 3452 d, 3181 d, 2873 d, 1711 f, 1611 f, 1590 m, 1512 m, 1449 m, 1324 m, 1308 m, 1252 m, 1209 m, 1183 m, 1173 m, 1121 m, 981 m, 959 m, 938 m, 831 m, 803 m, 777 m, 745 m, 683 d, 646 d. 1H-RMN (DMSO-d6,400 MHz): δ 1.70 (s, ancho, 4H, H8), 3.1 (s, ancho, 4H, H7), 3.90 (s, ancho, 4H, H6), 6.40 (m, ancho, 4H, H2' y H6'), 6.84 (s ancho, 2H, H1), 6.90 (d, J = 6.4 Hz, 2H, H3), 7.27 (m, ancho, 4H, H2 y H4), 7,50 (s, ancho, 2H, H5'), 8.77 (s, ancho, 2H, H4'), 9.66 (s, ancho, H1'). Λ (DMSO, 28 °C) (Ω-1.cm2.mol-1): 62.4.
4; polvo amarillo claro (rendimiento: 36 %). C37H37N4 O10Yb, porcentaje de iterbio Yb 21.3 ± 0.6 (calc. 20.2)%. p.f. > 300 °C. IR (ATR cm-1): и 3489 d, 3209 d, 2823 d, 1674 f, 1615 f, 1589 m, 1535 d, 1509 d, 1446 m, 1326 m, 1310 m, 1254 m, 1212m, 1176 d, 1123 m, 978 m, 951 m, 847 m, 800 m, 775 m, 748 m, 722 m, 686 m, 649 m. 1H-RMN (DMSO-d6, 400 MHz): δ 1.72 (s, 4H, H8), 3.13 (s, 4H, H7), 3.90 (s, 4H, H5), 4.34 (s, 4H, H6), 6.44 (m, ancho, 4H, H-2' y H6'), 6.87 (t, J = 7.4 Hz, 2H, H1), 6.92 (s, J = 8.1 Hz, 2H, H3), 7,30 (m, ancho, 4H, H2 y H4), 7.54 (m, ancho, 2H, H5'), 8.80 (s, 2H, H4'), 9.76 (s, ancho, H1 '). Λ (DMSO, 28 °C) (Ω- 1.cm2.mol-1): 56.9.
2.5 Determinación de parámetros de contraste
2.5.1 Relajatividad molar
La determinación de las relajatividades molares de los complejos lantánidos se llevó a cabo en un espectrofotómetro Bruker Ultrashield Avance II 400 operando a 400 MHz y con intensidad de campo de 9 T a 300 K. Se usó el medicamento DOTAREM® como agente de contraste de referencia en los estudios. La secuencia empleada para el ensayo es mostrada en la figura 5.
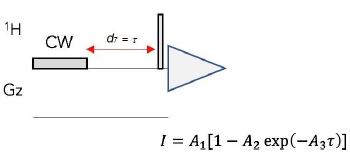
Figura 5 Secuencia "recuperación de saturación" para determinación de tiempos de relajación del agua (71 ) y ecuación de ajuste para cálculo de A3 = 1 /T1 [11].
Se realizó el cálculo de 71 para cada concentración presentada en la tabla 1, variando el parámetro (d7) tiempo de adquisición en 0 s, 0.25 s, 0.50 s, 1 s, 1.5 s, 2.0 s, 4.0 s, 6.0 s y 8.0 s.
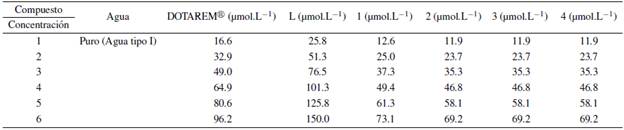
Las disoluciones fueron preparadas en 400 µL agua tipo I con 50 µL de agua deuterada (D2O). Posteriormente se realizó la gráfica de 1/T1 vs concentración de compuesto para determinar la relajatividad molar como la pendiente de la regresión lineal de los datos.
2.5.2 Rendimiento cuántico
Se determinaron los rendimientos cuánticos de los cuatro complejos lantánidos y el fluoróforo F. Se usó el espectrofluorímetro JASCO FP-8500 para la adquisición de los espectros de emisión y el espectrofotómetro UV-visible Evolution 220 Thermo Scientific para la toma de los espectros de absorción. Para cada compuesto se tomaron espectros de absorción de siete disoluciones de concentración variable de tal manera que las absorbancias variaron entre 0 y 0.1 unidades, evitando el fenómeno de autoabsorción al momento de adquisición de los espectros de emisión. Los resultados de rendimiento cuántico se expresan como relación de pendientes de las gráficas de emisión integrada vs absorbancia de los compuestos sobre el patrón, a su vez multiplicado por el rendimiento cuántico del mismo, como indica la ecuación 1 [12].
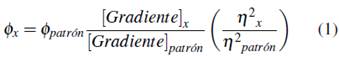
El patrón usado para estos experimentos fue fluoresceína en hidróxido de sodio 0.1 mol.L-1, cuyo rendimiento cuántico es 0.79 [8] y se empleó una longitud de onda de excitación de 370 nm.
2.5.3 Estabilidad termodinámica preliminar
La estabilidad termodinámica preliminar de los complejos se determinó a partir de la constante de equilibrio aparente de disoluciones 0.5 mol.L-1 de los complejos lantánidos y el medicamento de referencia, tras su incubación por 72 horas a 37 °C. Para ello se empleó la cuantificación de lantánidos libres por método colorimétrico usando naranja de xilenol. Se prepararon curvas de calibración para cada metal desde 1.0 µmol.L-1 hasta 5.0 µmol.L-1 usando el respectivo tricloruro de lantánido como patrón. Las muestras y curvas para lectura en el espectrofotómetro UV-visible Evolution 220 Thermo Scientific se prepararon en buffer de hexametilentetramina 50 mmol.L-1 ajustado a pH 7.0, el cual se empleó como solvente para preparar una solución de naranja de Xilenol 1.2 %. Las lecturas fueron tomadas por triplicado. Las constantes de equilibrio aparente (k ap ) fueron calculadas a través de la ecuación 2.
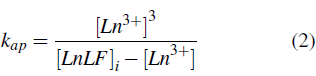
En donde k ap es la constante de equilibrio aparente; [Ln3+] es la concentración de lantánido libre y [LnLF] i es la concentración de complejo inicial.
2.6 Ensayos de susceptibilidad antimicrobiana
La determinación de la susceptibilidad antimicrobiana fue llevada a cabo mediante la obtención de las concentraciones mínimas inhibitorias (CMIs), siguiendo técnicas de microdilución estándar [13, 14]. Los complejos lantánidos y el ligando L fueron evaluados en seis cepas bacterianas ATCC, 3 Gram-positivas y 3 Gram-negativas: ATCC 25923 (Staphylococcus aureus), ATCC 10876 (Bacillus cereus), ATCC 19115 (Listeria monocytogenes), ATCC 25922 (Escherichia coli), ATCC 14028 (Salmonella typhimurium) y ATCC 27583 (Pseudomonas aeruginosa). Las microdiluciones se realizaron en placas de 96 pocillos partiendo de disoluciones 2000 mg.L-1 de los compuestos en caldo Muller-Hinton (CMH). Se llenó cada columna de la placa con 100 µL de CMH y se realizó la dilución con 100 µL de disolución de compuesto, pasando 100 µL de nueva disolución a la siguiente columna de la placa hasta la columna 11, finalmente se adicionaron 100 µL de inocuo bacteriano para completar los volúmenes hasta 200 µL. El rango de concentración evaluado estuvo entre 1000 mg.L-1 y 2 mg.L-1. El control negativo empleado fue CMH en ausencia de bacteria (primera columna de la placa) y el control positivo fue CMH con sólo inocuo de bacteria (última columna de la placa). Se utilizó ciprofloxacino clorhidrato como control de medicamento y nitrato de plata como referencia de inhibición.
2.7 Ensayos de interacción con el ADN
Se realizó una resuspensión en agua tipo I de ADN, para preparar una disolución de concentración 6820 µmol.L-1 en pares de bases, la cual fue determinada usando 6600 cm-1.mol-1.L como coeficiente de extinción molar y la absorbancia a 260 nm. Se obtuvo una relación de absorbancias A260/A280 de 1.88, indicando que el ácido nucleico preparado estaba libre de ARN y proteínas. Esta solución madre siempre se mantuvo almacenada a -5 °C.
2.7.1 Ensayos de absorción electrónica (UV-Vis)
Los espectros de absorción fueron medidos en el espectrofotómetro UV-visible Evolution 220 Thermo Scientific con accesorio Peltier para control de temperatura. Las titulaciones fotométricas se realizaron a una temperatura de 20 °C, a una concentración constante del compuesto y con adición de ADN cubriendo un rango desde 5 µmol.L-1 a 38 µmol.L-1. Las concentraciones del compuesto empleado para cada ensayo fueron L (42 pmol.L-1), F (37 µmol.L-1), 1 (25 pmol.L-1), 2 (24 pmol.L-1), 3 (23 µmol.L-1) y 4 (20 µmol.L-1). Tras la adición de cada alícuota de ADN, se tomó el respectivo espectro electrónico del compuesto estudiado en un rango de longitudes de onda de 230-550 nm con previa corrección de blanco. Para el cálculo de las constantes de unión, se ajustaron los datos obtenidos a la ecuación de Wolfe-Shimmer descrita en la ecuación 3.
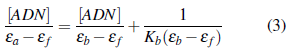
En dónde [ADN] es la concentración de ADN agregado, ε a es el coeficiente de extinción molar aparente, εb es el coeficiente de extinción molar totalmente unido y εf es el coeficiente de extinción molar del compuesto libre.
2.7.2 Mediciones de viscosidad
La viscosidad de seis soluciones de ADN a concentración de 600 µmol.L-1, con concentraciones incrementales de compuesto, fueron medidas con el uso de un semi-micro viscosímetro Cannon-Ubbelohde 75. Se evaluaron relaciones [ADN]/[Compuesto] de 0, 0.2, 0.5, 1.0, 1.5 y 2.0, manteniendo control de temperatura a 25 °C en un baño termostatizado. Los tiempos de flujo fueron medidos con un cronómetro.
3 Resultados y discusión
3.1 Análisis estructural
3.1.1 Espectroscopia de infrarrojo
La síntesis de los ligandos fue seguida a través de espectroscopia IR junto con la medición de algunos parámetros físicos para la confirmación de la estructura propuesta. Para el caso de L, se vigiló el avance de las reacciones teniendo en cuenta la presencia y ausencia de bandas características modificadas durante el esquema de síntesis. Las figura 6a y figura 6b muestran el seguimiento de las bandas características de cada compuesto intermedio para cada paso descrito en la figura 2.
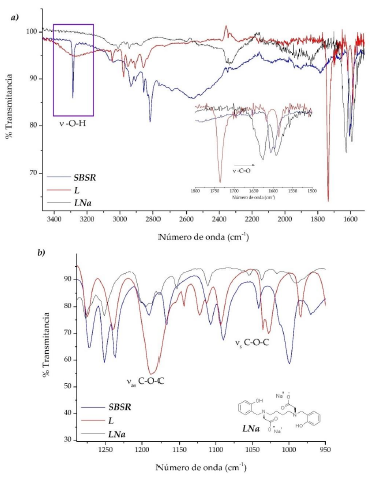
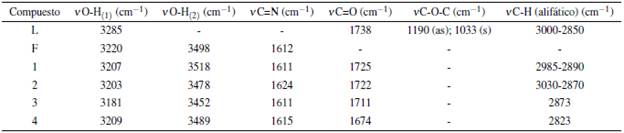
El ensanchamiento de la banda de tensión absorción de los grupos O-H aromáticos a 3300 cm-1, que ocurre al pasar del compuesto SBSR a L y que está ausente en LNa (compuesto hidrolizado por base) es la principal observación. Esta banda también se encuentra ausente en los espectros infrarrojos de todos los complejos, evidenciándose la tensión del grupo O-H del fluoróforo F. La desaparición de las dos bandas de tensión simétrica y asimétrica del enlace C-O-C característica de L en los demás espectros indica la efectiva hidrólisis del éster, además del desplazamiento de la banda de tensión del grupo carbonilo C=O hacia menor energía por disminución de efecto de resonancia ante la ausencia del grupo etilo. La comparación de los desplazamientos de las bandas características entre ligandos y el complejo se resumen en la tabla 2.
El posible modo de unión de los grupos carboxilo del ligando L hidrolizado hacia los centros metálicos, puede proponerse al analizar la diferencia de las frecuencias de tensión simétrica y asimétrica del enlace O-C-O. En la tabla 3 se resumen las frecuencias a evaluar junto con las diferencias entre ellas.
Tabla 3 Diferencias entre bandas de tensión simétrica y asimétrica del enlace O-C-O.
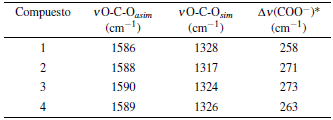
*Av = v O-C-Oasim - v O-C-Osim
Para el caso de los complejos lantánidos, se observó que la diferencias en frecuencias son mayores a 200 cm-1, sugiriendo una posible coordinación monodentada por parte los grupos carboxilatos del ligando L hidrolizado [14, 15].
De la tabla 2 se puede apreciar que, para las frecuencias de vibración de tensión de los grupos OH, se conserva únicamente la frecuencia de vibración correspondiente al fluoróforo F (v O-H(2)) en los complejos lantánidos, además de observarse un desplazamiento de aproximadamente 20 cm-1 en la banda de tensión del O-H adyacente al carbono ipso (v O-H(1)) que denota la posible coordinación a través de uno los O-H de los anillos bencénicos. En adición, no se observa desplazamiento significativo en las energías del enlace imino del fluoróforo F, indicando, probablemente, que el grupo azometino de la molécula no está participando en la coordinación [16].
3.1.2 Espectroscopia de resonancia magnética nuclear
El ligando L mostró las señales esperadas para la estructura propuesta, se diferencia de su posible contraparte sustituida en la posición 1 por el desplazamiento químico, que en este caso es de 10.02 ppm, característica de un protón en el grupo O-H aromático. En campo alto se observaron las señales de los protones alifáticos, correspondientes a los hidrógenos H9, H11, H10, H6, H7 y H8; los hidrógenos H9 y H8 se observaron como triplete que integra para seis hidrógenos y cuartete que integra para cuatro hidrógenos, respectivamente; los cuales se acoplan con constante J = 7.1 Hz; H6, H7 se observan como singletes que integran para cuatro hidrógenos, denotando la ausencia de protones adyacentes no equivalentes; H10 y H11 se encuentran como dos señales anchas sin resolución de acople, aunque se alcanza a observar que H11 pretende ser un quintete y H10 un triplete (ver material suplementario). En campo bajo se lograron observar las señales correspondientes a los hidrógenos aromáticos y aquellos enlazados a heteroátomos; los dobletes H2 y H5 que integran para dos protones se diferencian en que H2 se encuentra más protegido por efecto de resonancia del grupo OH bencénico, lo que hace que su desplazamiento químico sea a campo más alto respecto a H5; los tripletes H3 y H4 que integran para dos protones cada uno, se logran diferenciar por el mismo efecto de resonancia, haciendo que la posición para respecto al grupo OH (protones H4) se proteja más y la señal se ubique a un desplazamiento químico de mayor campo. Finalmente, para la región aromática sólo se observó acople meta entre los protones H3 y H5 y entre H2 y H4, con magnitudes de J = 0. 8 Hz y 0.7 Hz, respectivamente. El espectro RMN 1H de L con señales asignadas se muestra en
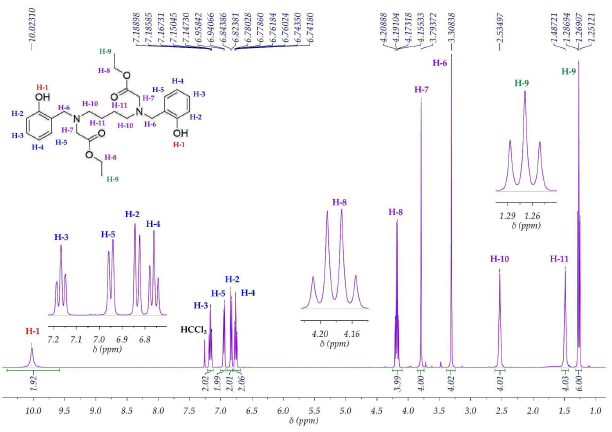
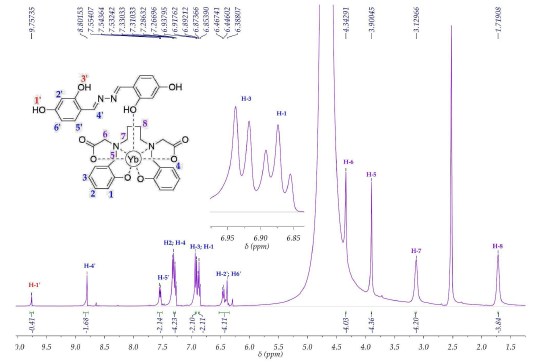
Para los complejos lantánidos, se observó la ausencia de las señales H9 y H8, indicando la hidrólisis efectiva del ligando L, además se encuentran las señales aromáticas del fluoróforo F y de algunos hidrógenos de heteroátomos, confirmando el anclaje de F al centro metálico. En adición, las señales se encuentran ensanchadas y modificadas en magnitud por el efecto paramagnético de los centros metálicos [17]; se presentaron también en general, desplazamientos hacia campo bajo, de las señales aromáticas correspondientes al ligando L hidrolizado y OH del fluoróforo F, indicando la posible coordinación a través de los grupos carboxilato e hidroxilo. La desaparición de la señal del protón H-3' del ligando F en todos los complejos indica la probable naturaleza intercambiable del protón en el grupo -OH coordinante, que en este caso podría ser el -OH en posición orto. En la información suplementaria se muestra el espectro 1H-RMN del ligando F.
De manera representativa, en la figura 8, se muestra el espectro 1H-RMN del complejo 4 con la asignación de señales, se realiza de manera similar para los complejos 1, 2 y 3 (ver material suplementario). Un aspecto particular de los espectros de los complejos, es el efecto del paramagnetismo del núcleo metálico en las señales de los ligandos. Se observa una disminución de las señales en los complejos más paramagnéticos (1 y 2) y un menor efecto en los complejos menos paramagnéticos 3 y 4.
3.2 Análisis físicos
3.2.1 Espectrometría de masas
Para los ligandos L y F se adquirieron espectros de masas que corroboraron la estructura propuesta.
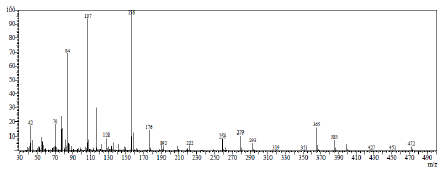
En la figura 9 se muestran los fragmentos característicos de L; se observó un fragmento característico, además del ion molecular a m/z = 472, que distingue la sustitución en NH en lugar del OH en el paso de SBSR mostrado en la figura 2, es el fragmento a m/z = 365, que indica la ruptura del enlace N-C6, además de la generación del fragmento m/z = 107 de alta intensidad que corresponde al fragmento fenólico sin sustitución.
En el caso del ligando F (ver material suplementario), el espectro de masas también presenta fragmentos característicos de compuesto, además del ion molecular con alta intensidad en m/z 272. Los iones de mayor intensidad corresponden a los fragmentos generados por la pérdida de un OH (m/z = 255), ruptura del enlace entre carbono ipso y C4' (m/z = 109), ruptura de enlace imino (m/z = 122) y ruptura simétrica del enlace N-N (m/z = 136, 137 y 138).
Los puntos de fusión encontrados para todos los compuestos fueron altos, para el caso de los complejos lantánidos, todos superaron los 300 °C; los análisis de conductividad molar muestran que podrían presentarse como compuestos iónicos, ya que los valores de conductividad superan los 50 Ω - 1 .cm2.mol-1, siendo similares a aquellos obtenidos para el yoduro de potasio en DMSO en condiciones similares [18, 19]. Es probable que los protones de los grupos hidroxilo coordinantes en las estructuras propuestas sean protones ácidos, pues al disolverse en agua logran bajar el pH del sistema hasta 6, y en medio básico también logran mantener su solubilidad, esto explica las altas conductividades encontradas para estos complejos, además de la ausencia de estos protones en los espectros de 1H RMN, que se piensa es debida a intercambio con solvente deuterado.
Los complejos de coordinación fueron parcialmente solubles en agua, y aumentaron su solubilidad en medios ácidos y básicos. En contraste, los ligandos L y F no fueron solubles en agua, pero sí en disolventes orgánicos como diclorometano, cloroformo, acetonitrilo y metanol. La solubilidad del ligando L aumenta en agua ligeramente acidulada (pH 5-6), gracias -posiblemente- a los nitrógenos aceptores de protones que le logran conferir carga neta a la molécula.
El porcentaje de lantánido en cada complejo fue determinado a través de la digestión ácida, y la posterior cuantificación colorimétrica de lantánidos libres, de acuerdo con la metodología seguida por Barge et al. [7]. Los porcentajes encontrados concuerdan con los valores teóricos para las posibles estructuras propuestas.
3.2.2 Análisis termogravimétricos
La figura 10 muestra el termograma del compuesto 4 como representativo de los cuatro complejos obtenidos.
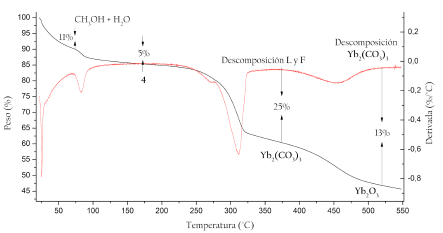
Se logró observar una pérdida de masa inicial en dos etapas, la primera a 50 °C (11 %) y la segunda a 95 °C (5 %), probablemente atribuidas al metanol y la humedad residual después de los pasos de recristalización y lavados. No se observan pérdidas entre 100 ° C y 120 ° C características de aguas de esfera de coordinación interna. Posteriormente se observa una pérdida hasta el 61 % de la masa inicial cuando se alcanzan los 312 °C, atribuida a una probable descomposición de los ligandos orgánicos, dejando como intermediario el carbonato de iterbio(III) (Yb2(CO3)2), finalmente se pierde otro porcentaje de masa hasta alcanzar el 47 % de la masa inicial, debida a la pérdida de monóxido de carbono (CO) producto de la descomposición del carbonato de iterbio, dejando finalmente el óxido de iterbio(III) (Yb2O3) como producto final de la descomposición térmica. Se tienen reportes de esta misma tendencia de descomposición para los complejos lantánidos unidos a ligandos con grupos carboxilatos y oxigenados, en donde los pasos finales se relacionan con la formación del respectivo carbonato de lantánido y su posterior paso a óxido [14].
Se obtuvieron termogramas similares para los complejos 1, 2 y 3, con la diferencia de no hacerse distinguible la última temperatura del paso desde carbonato de lantánido a óxido (ver material suplementario). Los termogramas mostraron coincidencias entre los porcentajes esperados de las especies intermediarias en la descomposición térmica y los obtenidos experimentalmente, teniendo en cuenta las masas molares de las estructuras moleculares propuestas.
3.3 Parámetros de contraste
Los parámetros físicos, evaluados de manera preliminar, para determinar los potenciales usos de los compuestos lantánidos obtenidos en aplicaciones diagnósticas fueron realizados de manera in-vitro sin emplear ningún modelo biológico.
3.3.1 Relajatividad molar
El funcionamiento de los agentes de contraste en resonancia magnética nuclear de imágenes, se basa en la modificación de los tiempos de relajación de los espines nucleares de los átomos de hidrógeno en el agua, de tal manera que, si los compuestos logran modificar en un mayor grado los tiempos T 1 o T2, se considerarán mejores agentes de contraste [6, 20 - 22].
Se determinaron las relajatividades molares en T1 (r1) de los cuatro complejos obtenidos y del compuesto de referencia Dotarem®, el cuál es usado comercialmente como medicamento para contraste en resonancia nuclear de imágenes (RMI). Además, se evaluó el efecto del ligando L en el tiempo de relajación del agua a través de r1 con un fin comparativo. Con el uso de la secuencia "Recuperación de saturación" se obtienen las curvas de intensidad de señal vs tiempo d7 de las cuales se obtiene el parámetro T1 para cada concentración (ver figura 11).
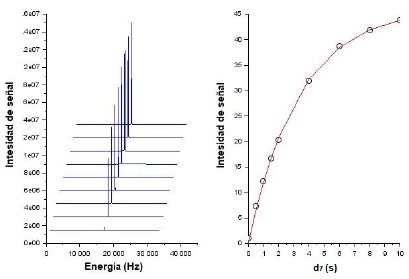
Figura 11 Recuperación de saturación en señales del agua y ajustes matemáticos para determinación de T1 del agua tipo I.
La figura 12 muestra el resumen de las seis rectas experimentales generadas a partir de la relación 1/T1 vs concentración. Los tiempos T1 obtenidos para cada compuesto se muestran en el material suplementario. En este experimento se obtuvo un T1 del agua tipo 1 de 3.10 s, que coincide con algunos valores reportados en la literatura [23, 24].
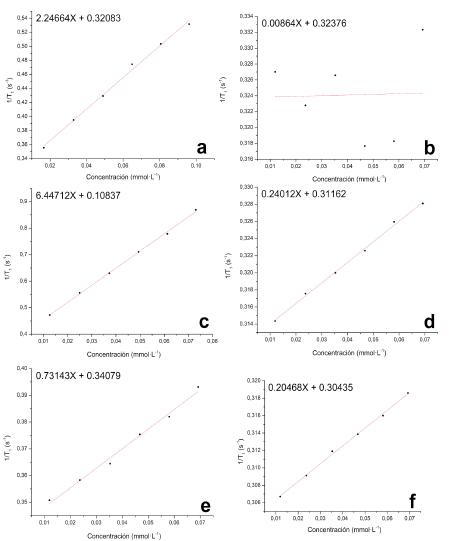
Figura 12 Curvas de relajatividad molar (r1) para compuestos evaluados en agua tipo I, 9 T y 300 K a). Dotarem® ; b). L; c). 1;d). 2;e). 3 y f). 4.
En la tabla 4 se resumen los resultados de r1 y en la figura 32 se observa una gráfica comparativa de las rectas para los seis compuestos.
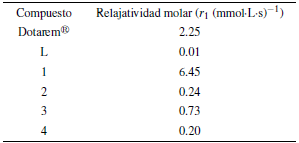
Se observó un aumento significativo en el parámetro r1 para el compuesto 1 inclusive por encima del compuesto de referencia Dotarem®. El efecto es concordante con el menor número de coordinación del metal respecto al compuesto de referencia, en este caso 7 para el compuesto 1 y 8 para Dotarem®, que permite que se tengan más aguas de coordinación en el complejo en solución y así el efecto paramagnético sobre los espines nucleares sea mayor [6, 20 - 22, 25]. Además, el tiempo de intercambio de aguas tm puede ser mayor debido a la mayor accesibilidad que tienen las moléculas de agua en un ligando lineal [26]. Por otro lado, el tiempo de correlación tr no debería aumentar, pues no hay presencia de macromoléculas que vuelva más lento el tambaleo de la magnetización sectorial que provoca el centro metálico paramagnético, por lo que se puede afirmar, que la principal causa del mejoramiento de r1 posiblemente sea el menor número de coordinación y la mayor tasa de intercambio de aguas entre esferas internas de coordinación y el seno del sistema (tm) [6, 26].
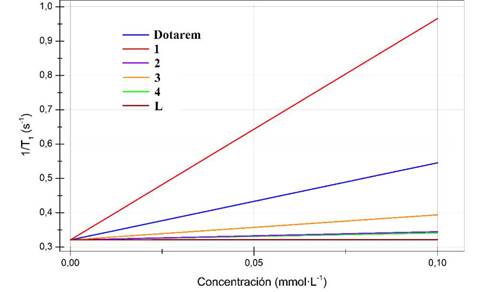
Los valores de r1 para los compuestos 2, 3, 4 y L estuvieron por debajo del compuesto de referencia Dotarem® y de 1 debido a su menor paramagnetismo. El paramagnetismo de los metales lantánidos dependerá en gran medida de su estado de oxidación y de cómo se desdoblan los orbitales f en el arreglo geométrico de los orbitales moleculares [6]. A pesar que el ión Eu(III) posee una configuración electrónica [Xe]4f6 en su forma de ion libre, en donde 6 electrones en los orbitales 4 f estarían desapareados, para el caso de 2, se podría tener un arreglo de orbitales en donde no todos los 6 electrones están desapareados, y así se reduzca en gran medida el paramagnetismo del ion, desembocando en un efecto poco importante sobre la modificación de los tiempos de relajación del agua [6, 25].
Los compuestos 3 y 4 tampoco presentan un significativo r1 en contraste con el compuesto de referencia Dotarem®, en este caso, por su reducido paramagnetismo al poseer mayor cantidad de electrones en sus orbitales f. Aun así, el compuesto 3 logró tener mayor efecto en r1 que el complejo 2. El ligando L mostró un efecto casi nulo sobre r1, confirmando su naturaleza diamagnética.
Las relajatividades molares, en este caso r1, son sensibles a la temperatura, viscosidad, intensidad del campo, entre otras, por lo que la comparación de datos con la literatura se debe hacer de manera cuidadosa[6], [23 - 25]. Por este motivo, las comparaciones se realizan con el compuesto de referencia Dotarem® bajo las mismas condiciones de viscosidad, temperatura, intensidad del campo. En todos los complejos se observó una aceleración de los tiempos de relajación del agua, lo que indica que pueden ser potencialmente usados como contrastes en resonancia magnética nuclear de imágenes (RMI), especialmente el compuesto 1.
3.3.2 Rendimiento cuántico de fluorescencia
La figura 14 muestra los espectros de emisión y absorción de la fluoresceína y el compuesto 4, los demás espectros se pueden encontrar en el material suplementario.
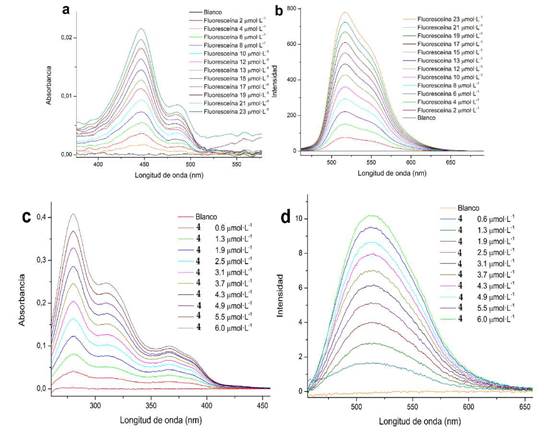
Figura 14 Espectros de absorción y emisión para determinación de rendimiento cuántico en NaOH 0.1 M. a). Absorción de Fluoresceína; b). Emisión de Fluoresceína; c). Absorción de 4 y d). Emisión de 4.
Se mantuvo una absorbancia < 0.1 en todas las disoluciones usadas para la determinación del rendimiento cuántico, con el fin de evitar fenómenos de autoabsorción; se realizó la determinación de las pendientes de las curvas "absorbancia vs intensidad integrada" para todos los compuestos evaluados. La figura 15 muestra las rectas generadas. El rendimiento cuántico de la fluoresceína es de 0.79 bajo las condiciones en que se realizaron los ensayos [12, 27], todos los experimentos se realizaron en el mismo disolvente, por lo que la razón de los coeficientes de refracción (n) de la ecuación 1 se considera 1. La tabla 5 muestra los valores de rendimiento cuántico respecto a la fluoresceína obtenidos para los compuestos evaluados.
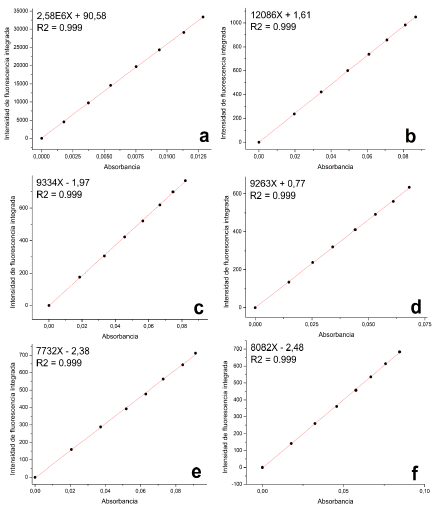
Figura 15 Curvas de intensidad de fluorescencia integrada vs absorbancia: a). Fluoresceina; b). F; c). 1; d). 2; e). 3 y f). 4.
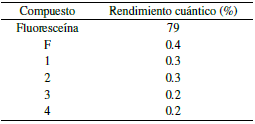
Todos los rendimientos cuánticos obtenidos están por debajo del 0.5 %, indicando que el proceso de emisión de fotones absorbidos es ineficiente en comparación con la fluoresceína. Se observa que el fluoróforo F al acoplarse con los centros metálicos disminuye su capacidad de emitir fotones, es decir se genera un leve quenching de la radiación, probablemente por la aparente coordinación directa al centro metálico a través del grupo OH, que podría inhibir los procesos de transferencia de energía [26, 9, 10], Sin embargo, los rendimientos cuánticos obtenidos se encuentran dentro del rango de algunos agentes de contraste usados para microscopía de fluorescencia [6, 28]. La figura 16 muestra la comparación de los gradientes (pendientes de regresión lineal) del fluoróforo F y los complejos lantánidos.
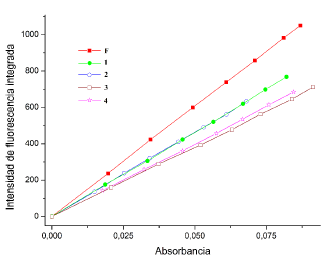
Se observa que el quenching es mayor para los complejos con centros lantánidos más pesados (Dy e Yb) en comparación con los más livianos (Gd y Eu). Es posible que el menor tamaño de los iones más pesados, que se traduce en una mayor carga nuclear efectiva, altera de manera más significativa las energías de los estados singlete y triplete, generando una diferencia de energía mayor y ocasionando que el entrecruzamiento de sistemas y la óptima transferencia de energía para la emisión radiativa, se vea disminuida [6]. Además, en contraste con los resultados de relajatividad molar, es probable que las aguas de coordinación presentes en solución y/o algunos iones OH provoquen el quenching en todos los complejos [6]. La proximidad o la unión directa de la antena F para la recolección de energía, no logra verse reflejada de manera totalmente beneficiosa en la región del espectro visible, sin embargo, para la región del infrarrojo e infrarrojo cercano, se tiene evidencia que los iones lantánidos como el Eu(III) poseen excelentes rendimientos, de tal manera que podría evaluarse la emisión en esta región del espectro [6, 29, 30].
3.3.3 Estabilidad termodinámica preliminar
Se pretendió emular las condiciones fisiológicas en pH 7.0 y temneratura 37 °C durante un neriodo de 72 horas (144 veces más, que la vida media del medicamento Dotarem® en el cuerpo y 7 veces más, que la vida media del Magnevist®) [31 - 33] para cuantificar posteriormente la cantidad de iones lantánidos libres y estimar la constante de estabilidad termodinámica de manera preliminar a través de la ecuación 2. La figura 17 muestra la curva de calibración empleada para la cuantificación de gadolinio libre, de manera análoga se obtuvieron curvas de calibración para los demás metales (ver material suplementario).
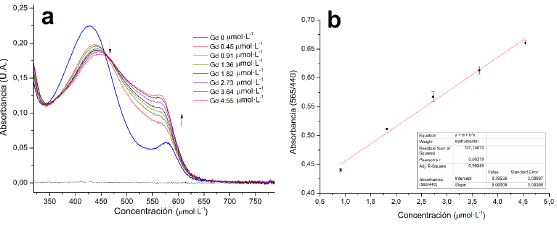
Figura 17 Cuantificación espectrofotométrica de lantánidos libres a). Espectros UV-Vis y b). Curva de calibración del Gd3+ libre.
La tabla 6 muestra los valores de - log [k ap ] encontrados después de la incubación y determinación por el método espectrofotométrico.
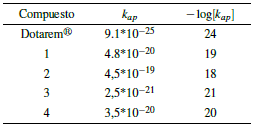
En este caso, la constante de estabilidad se asume como el inverso de la constante de disociación de los complejos (kap). Todos los complejos tuvieron estabilidades menores a las del compuesto de referencia Dotarem®, entre 5 y 6 órdenes de magnitud por debajo, lo cual reafirma la mayor estabilidad termodinámica que confiere los ligandos macrocíclicos sobre los polidentados lineales, sin embargo, se obtuvieron constantes altas en referencia a constantes halladas por técnicas similares para otros ligandos polidentados y agentes de contraste [5, 6] entre los complejos; se observa una tendencia de incrementar la estabilidad en cuanto se disminuye el tamaño del centro metálico en 1 o 2 órdenes de magnitud, probablemente por el aumento de la dureza del ion que lo convierte en una especie más oxófila [5, 34].
Las altas estabilidades encontradas a los tiempos de incubación trabajados señalan a los compuestos obtenidos como posibles candidatos viables para contraste tanto en resonancia como en microscopía de fluorescencia, ya que en general los metales lantánidos y sus complejos no son retenidos en el cuerpo por más 24 horas [33]. Además, la alta relajatividad molar y relativa alta estabilidad del compuesto 1 puede desembocar en su uso a menores dosis para generar el mismo efecto en el mejoramiento de imágenes T1 [6], [35, 36].
3.4 Susceptibilidad antimicrobiana
Se evaluó la susceptibilidad de seis cepas bacterianas ATCC frente a los compuestos L, 1, 2, 3 y 4 por el método de microdilución. La tabla 7 resume las concentraciones mínimas inhibitorias (CMIs) encontradas para las diferentes cepas y diferentes compuestos. El ciprofloxacin) clorhidrato (Cip) y el nitrato de plata (AgNO3) son usados como controles de inhibición.
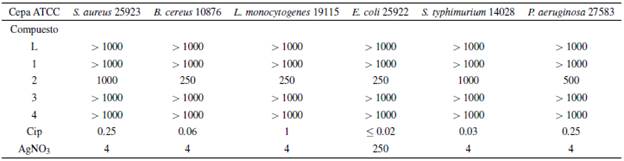
Se encontró en general que tanto el ligando L como los complejos lantánidos no inhiben el crecimiento de las cepas bacterianas evaluadas a las concentraciones de los controles. Estos resultados posiblemente sean consecuencia de la ausencia de un fuerte farmacóforo en las estructuras moleculares propuestas para los complejos lantánidos, siendo el fluoróforo F la única entidad con actividad biológica de acuerdo a la literatura, sin embargo, no es suficientemente eficiente para inhibir el crecimiento bacteriano a las concentraciones evaluadas [9, 10]. El compuesto con mayor actividad entre los complejos lantánidos fue 2, con CIMs desde 250 mg-L-1 hasta 1000 mg-L-1, centrándose la efectividad de la inhibición en cepas Gram-positivas. La mayor actividad encontrada para 2 podría estar relacionada con un posible aumento de la lipoficidad por la disminución del tamaño del ion lantánido, que logra afectar más fácilmente las bacterias Gram-positivas[14]. De esta manera, gracias a que los complejos no logran inhibir el crecimiento de algunas cepas bacterianas a concentraciones relativamente altas, podrían tener una potencial aplicación como marcadores fluorescentes para estudios in-vitro de células patógenas en concentraciones menores a las CIMs, esto se podría corroborar a través de ensayos de tinción y lectura por microscopía de fluorescencia.
3.5 Ensayos de interacción con el ADN
La forma en que interaccionan los compuestos obtenidos con las hebras bicatenarias del ADN pueden dar un indicio de la citotoxicidad a nivel de los procesos de replicación y transcripción; así, los resultados que se obtienen de estos experimentos pueden dar cuenta de algunos posibles mecanismos de acción en efectos biocidas, mecanismos de marcaje biológico y mecanismos de citotoxicidad no deseada [14], [19], [37]. Se estudió el posible modo de unión de los complejos lantánidos obtenidos, el cual fue comparado contra el ligando L y el fluoróforo F, a través de la estimación de las constantes de unión K b .
3.5.1 Ensayos de absorción electrónica
En primer lugar, se realizaron las titulaciones espectrofotométricas con ADN de los ligandos empleados (L y F) para la formación de los complejos (figura 18).
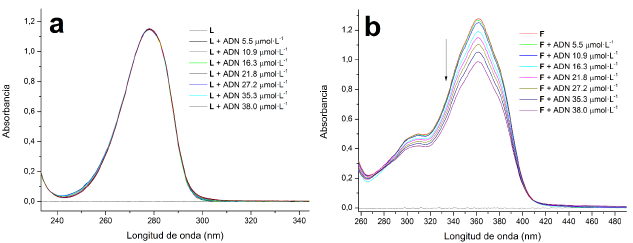
Figura 18 Efecto de la adición de ADN de timo de ternera sobre el espectro electrónico de a). L y b). F.
En el espectro electrónico de L (figura 18a), no se observa un efecto neto sobre la banda de absorción cerca de los 280 nm, mientras que el espectro electrónico del fluoróforo F (figura 18b) se evidencia un efecto hipocrómico en su banda de absorción cerca de los 360 nm, indicando una posible interacción por intercalación entre F y las pares de bases del ADN [14, 19, 37]. La explicación puede residir en que el fluoróforo F posee una estructura conjugada más extensa y por ende una mayor planaridad, lo que lo hace poseer un sistema n extenso que logra estabilizar las bases nitrogenadas por interacción de apilamiento n [19], propiedad que no posee el ligando L. La figura 19 muestra el efecto de la interacción de los complejos lantánidos con el ADN.
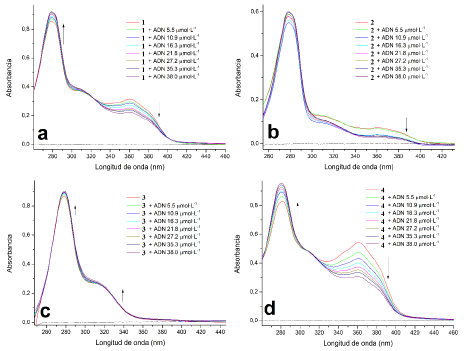
Figura 19 Efecto de la adicion de ADN de timo de ternera sobre el espectro electronico de a). 1; b). 2; c). 3 y d). 4.
En los espectros electrónicos de los complejos 1, 2 y 4, se observaron cambios hipocrómicos en la banda de absorción cerca de 360 nm e hipercrómicos en la banda cerca de los 280 nm, a medida que aumenta la presencia del ADN en el sistema; lo cual indica una posible interacción entre los complejos metálicos y las hebras del ADN. Además, se observan también puntos isosbésticos durante la titulación que indican la formación de una tercera especie, posiblemente la especie ADN-Complejo [14,37]. Se identifica de manera general, que las bandas características de los complejos se comportan de manera distinta; por ejemplo, la banda a 360 nm que presenta hipocromismo, y que principalmente pertenece al fluoróforo F, puede estar indicando que su coordinación al centro metálico de los complejos favorece su intercalación con el ADN, a través de ese fragmento de la molécula; el efecto hipercrómico en la banda a 280 nm, principalmente asociada al ligando hidrolizado L, indica que también se ve afectada la absorción al ocurrir el proceso de intercalación, efecto que no se observa en el ligando L libre. De esta manera, la interacción con las hebras del ADN de los complejos lantánidos puede estarse dando posiblemente por un mecanismo de inserción, que implica tanto la intercalación entre las bases nitrogenadas del ADN, como la unión a los surcos del esqueleto de fosfatos [19]. Por otro lado, el efecto neto sobre el espectro electrónico del 3 fue el leve hipocromismo en ambas bandas de absorción, lo que sugiere también una posible interacción con las hebras del ADN, sin involucrar precisamente el proceso de intercalación [37].
Con los datos de absorbancia de la banda a 360 nm de la figura 19 y las concentraciones de ADN empleadas, se ajustaron los datos a la ecuación 3 y se usaron los valores de pendiente e intercepto para el cálculo de las constantes de unión. La tabla 8 presenta los valores de las constantes de unión encontradas para los compuestos evaluados.
Los valores encontrados para las constantes de unión son menores a las reportadas para intercaladores clásicos como el bromuro de etidio, entre otros, las cuales se encuentran en el rango de 105-106 L-mol-1 [9, 37]. La constante de unión estimada para el complejo 3 es dos órdenes de magnitud más baja que el resto de complejos. En general, estas constantes de unión dan cuenta de la posible toxicidad a nivel genómico que tienen los complejos lantánidos [19], el hecho que los valores kb sean menores que aquellos para intercaladores clásicos como el bromuro de etidio, que se consideran altamente tóxicos y mutagénicos, puede dar indicios de una toxicidad reducida, al menos, al nivel de los procesos de replicación y transcripción de ácidos nucleicos. La toxicidad que pudieran presentar éstos complejos lantánidos a través de otros mecanismos fisiológicos, debe ser evaluada por medio de otros ensayos y experimentos de citotoxicidad in-vitro e in-vivo.
3.5.2 Mediciones de viscosidad
Los cambios en la viscosidad de una solución de ADN dan cuenta de la forma en que puede interaccionar las moléculas con las hebras de los ácidos nucleicos [14], [19], [37]. La figura 21 muestra los resultados del efecto de los compuestos sobre la viscosidad relativa de una solución de ADN 600 mmol-L-1.
Las viscosidades relativas del ADN en general incrementaron a medida que se aumentó la relación [Compuesto]/[ADN], con excepción del ligando L. Esto indica nuevamente que, posiblemente, la forma de unión con las hebras del ADN sea a través del mecanismo intercalativo, ya que la inserción de las moléculas entre las bases nitrogenadas del ADN aumenta la longitud del biopolímero, teniendo como efecto un aumento de la viscosidad del mismo a nivel macroscópico [14, 19]. También hay congruencia en que el ligando L no tiene la capacidad de unirse al ADN, pues se observó que la viscosidad relativa se mantuvo casi constante en todo el rango de ratios evaluados, confirmando lo observado en los ensayos de seguimiento del espectro electrónico.
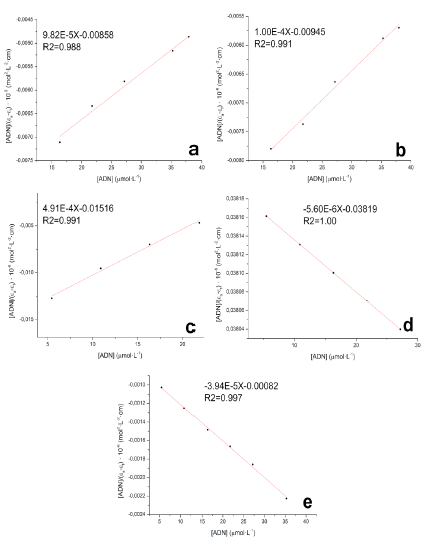
Figura 20 Ajustes de Wolfe-Shimmer para determinación de constantes de unión K b de los compuestos a). F; b). 1; c). 2; d). 3 y e). 4.
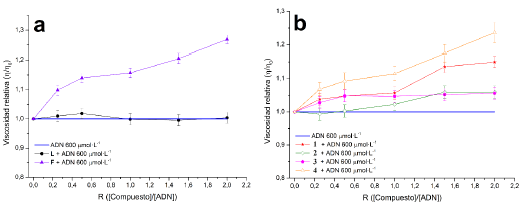
Figura 21 Efecto en la viscosidad relativa del ADN en presencia de los compuestos a). L y F; b). 1, 2, 3 y 4.
De esta manera, los compuestos obtenidos mostraron una posible capacidad de unirse a las hebras del ADN, de tal manera que éstos podrían intervenir, de manera reducida, en los procesos bioquímicos que involucran a los ácidos nucleicos, como los procesos de replicación y transcripción. Esto se podría correlacionar con la posible baja citotoxicidad que pudieran presentar los complejos lantánidos de manera in-vitro, además de poder explicar la reducida actividad biocida contra células procariotas y su potencial aplicación para contraste óptico y contraste para RMI intravenoso.
4 Conclusiones
Se obtuvieron una serie de complejos lantánidos con iones Gd(III), Eu(III), Dy(III) e Yb(III) a partir de un ligando polidentado y un fluoróforo, los cuales fueron caracterizados a través de técnicas espectroscópicas y físicas. Las estructuras propuestas para los complejos lantánidos estuvieron en concordancia con los análisis de caracterización estructural. Dentro de los parámetros preliminares evaluados para estudiar el potencial uso de estos compuestos para aplicaciones diagnósticas en imagenología, se concluye que todos los complejos lantánidos evaluados logran modificar el tiempo de relajación del agua, encontrándose el mayor efecto en el complejo 1, que incluso fue casi tres veces mayor que el compuesto de Dotarem®, usado comercialmente para diagnóstico clínico. Los rendimientos cuánticos encontrados mostraron que la eficiencia con la que el fluoróforo F emite la radiación absorbida en radiación visible, es muy baja en comparación con la eficiencia de la fluoresceína; el acomplejamiento del fluoróforo a los centros metálicos, provocó una disminución en la emisión de radiación; sin embargo, es posible evaluar su potencial uso como agente de contraste celular en células procariotas, teniendo en cuenta los valores de CMI obtenidas. Las estabilidades termodinámicas preliminares indicaron que los complejos logran alcanzar una estabilidad de hasta cuatro órdenes de magnitud por debajo del medicamento de referencia a las condiciones evaluadas, indicando una posible viabilidad para su uso. Los resultados mostraron que los compuestos tienen un posible potencial para ser empleados en resonancia magnética nuclear de imágenes, especialmente el compuesto 1, cuya relajatividad molar r1 fue mayor que el medicamento de referencia, el cual podría reducir las dosis requeridas para generar un mejoramiento en el contraste de imágenes tipo T1. Finalmente, los ensayos de interacción con el ADN indicaron que los compuestos obtenidos podrían interaccionar con las hebras bicatenarias, teniendo posibilidad de intervenir en los procesos de transcripción y replicación, en este caso, si los compuestos llegaran a encontrarse directamente con el material genético de una célula.

