










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Reumatología
Print version ISSN 0121-8123
Rev.Colomb.Reumatol. vol.14 no.2 Bogotá Apr./June 2007
Artículo de Revisión
Enfermedad pulmonar intersticial asociada a enfermedades de tejido conectivo
An interstitial lung disease (ILD) belongs to a group of diffuse parenchymal lung diseases.
Yimy F. Medina1, José Félix Restrepo2, Antonio Iglesias3, Paulina Ojeda4, Carlos Matiz5
1 Médico Internista y Reumatólogo. Universidad Nacional de Colombia.
2 Profesor Titular de Medicina Interna y Reumatología Universidad Nacional de Colombia. Coordinador Unidad de Reumatología.
3 Profesor Titular de Medicina Interna y Reumatología Universidad Nacional de Colombia.
4 Patóloga Hospital Santa Clara. Profesora Universidad El Bosque.
5 Neumólogo Clínica El Bosque. Profesor Asistente Universidad El Bosque.
Recibido para publicación: marzo 29/2007 Aceptado en forma revisada: mayo 31/2007
Resumen
La enfermedad pulmonar intersticial pertenece al grupo de la enfermedad pulmonar parenqui-matosa difusa. Debe ser diferenciada de otras patologías entre las que están las neumonías intersticiales (NII) asociadas a enfermedad de tejido conectivo (ETC) y las idiopáticas. Se han originado nuevos conceptos en los últimos años y se las ha clasificado en siete subgrupos bien definidos y se ha descrito la asociación de cada uno de estos subgrupos con las ETC. Su historia natural y otros aspectos de su tratamiento no se conocen completamente. Para su diagnóstico completo se requieren criterios clínicos, imagenológicos y de histopatología. La biopsia pulmonar ocupa un lugar esencial. Es importante promover y estimular la subclasificación de cada subgrupo con el fin de conocer su historia natural, dirigir el tratamiento y mejorar su pronóstico.
Palabras clave: enfermedad pulmonar intertisticial, enfermedad del tejido conectivo.
Summary
It should be differentiated from other pathologies among those are idiopathic and ILD associated to connective tissue diseases (CTD). New concepts have been developed in the last years, and they have been classified in seven defined subgroups. It has been described the association of each one of these subgroups with CTD. Natural History and other aspects of its treatment is not known completely. For complete diagnose it is required clinical, image, and histopathologic approaches. The biopsy lung plays an essential role. It is important to promote and to stimulate the subclasification of each subgroup with the purpose of, knowing their natural history, directing the treatment and to improve their outcome.
Key words: interstitial lung disease, connective tissue diseases.
La enfermedad pulmonar intersticial (EPI) o neumonía intersticial idiopática (NII) es un grupo heterogéneo de enfermedades no neoplásicas que resultan del daño del parénquima pulmonar mediante varios patrones de inflamación y fibrosis. Como su nombre lo indica, estos desórdenes tienen como sitio primario de patología y daño, el intersticio. El intersticio comprende el espacio entre el epitelio y el endotelio de las membranas. Con frecuencia se ven afectados, además del intersticio, los espacios aéreos, las vías aéreas periféricas y los vasos junto con sus respectivos recubrimientos epiteliales y endoteliales1.
La enfermedad pulmonar parenquimatosa difusa (EPPD) la componen diferentes entidades de causas conocidas y desconocidas en las que están incluidas las neumonías intersticiales idiopáticas (Ver Tabla 1), tema en el que nos vamos a centrar en este artículo. Las NII deben ser diferenciadas de otras causas de EPPD, particularmente de las drogas y de la sarcoi-dosis, pero también de otras neumonías intersticiales de causas conocidas como las NII asociadas a enfermedad de tejido conectivo (ETC).

Se han originado conceptos nuevos en la enfermedad intersticial pulmonar (otros sinónimos son fibrosis pulmonar, neumonía intersticial idiopática, enfermedad pulmonar parenquimatosa y que en Estados Unidos se le ha llamado fibrosis pulmonar idiopática, en Inglaterra neumonía intersticial y en Japón alveolitis fibrosante criptogénica) especialmente desde que se realizó el consenso de la Sociedad Americana de Tórax y la Sociedad Respiratoria Europea2 (SAT/SRE) que definió un grupo de patrones histológicos con el fin de suministrar las bases para un diagnóstico clínico radiológico patológico definitivo de las NII.
Los subtipos de fibrosis pulmonar o neumonías intersticiales que se han propuesto son los que están enumerados en la Tabla 2. Es importante tener claro que se ha recomendado el uso de los diferentes términos de acuerdo a si la denominación tiene en cuenta la histopatología o la clínica; por ejemplo, con frecuencia se hace referencia a la entidad neumonía intersticial usual (NIU), pero este es un término histopatológico, por tanto se debería hablar de la fibrosis pulmonar idiopática y cuando se haga referencia a la histopatología o a imágenes referirse al término de NIU e idealmente adicionar el término "patrón". Así, por ejemplo, cuando en la bronquiolitis respiratoria asociada a EPID que es la entidad clínica debida a diversas causas y cuando hablamos de la descripción histopatológica, hablamos de patrón de bronquiolitis respiratoria asociada a EPI.
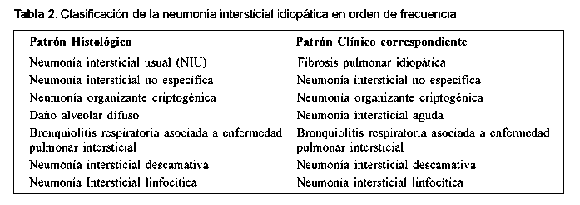
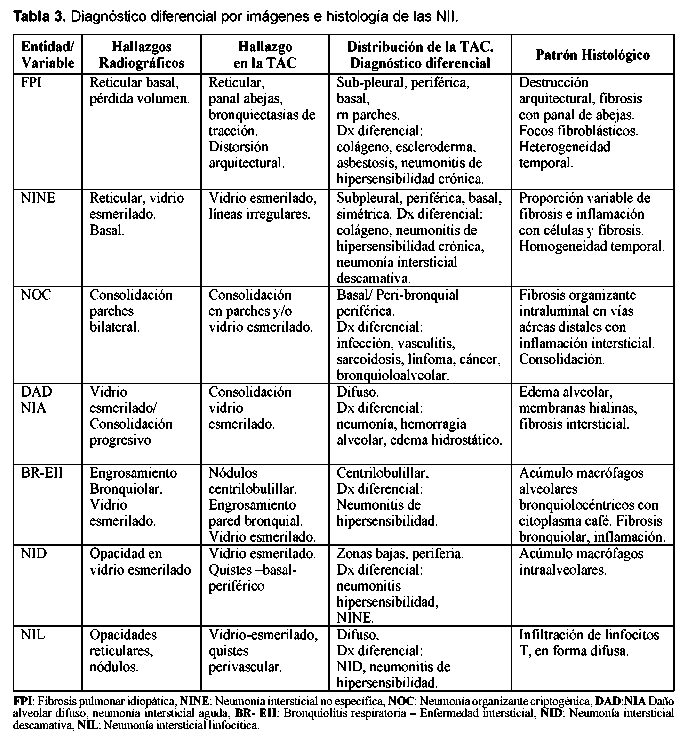
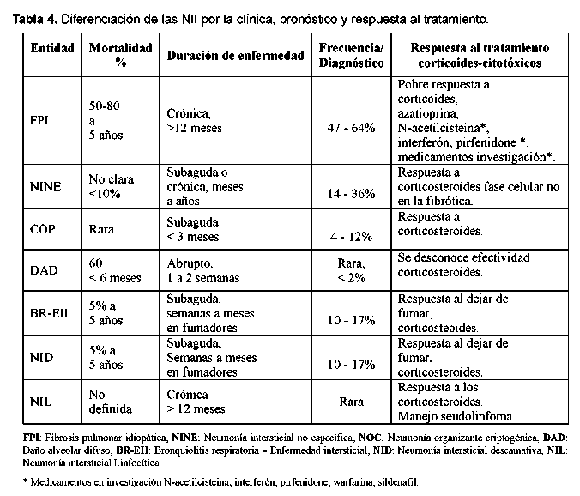
A pesar de que se considera a las enfermedades intersticiales pulmonares idiopáticas un grupo diferente a las neumonías intersticiales causadas por las enfermedades de tejido conectivo, se ha visto que el daño pulmonar es idéntico en los dos grupos y por El diagnóstico diferencial de las EPI asociadas a esto se ha presumido que el tratamiento y el pronós- ETC se hace principalmente con las otras causas de tico sean igualmente similares3. Este concepto no es la EPDD (Tabla1), pero también un enfoque impor-aceptado completamente ya que existen pocos estu- tante en su diferenciación es distinguirla de las otras dios de la enfermedad natural de la EPI en las ETC. NII principalmente porque se ha considerado que la fibrosis pulmonar idiopática responde escasamente a los corticoides a diferencia de las otras seis entidades de la EPI que tienen una respuesta más favorable4 (Ver Tablas 3 y 4). Algunas infecciones pueden simular la EPI. Una infección que requiere especial atención es la infección por Pneumocystis carinii5 que se ha visto que tiene predilección en pacientes con lupus eritematoso sistémico6, en pacientes con miopatías inflamatorias7, uso de corticoides, metotrexate8 o inhibidores del factor de necrosis tumoral alfa9 y que cuando está presente se debe tener también en cuenta para el diagnóstico diferenciado de la NII.
Fibrosis pulmonar idiopática / Neumonía intersticial usual (NIU)
Es la forma más común de las neumonías intersticiales idiopáticas10. Los síntomas suelen ser graduales con tos seca paroxística y disnea en pacientes mayores de 50 años y ligeramente predominante en los varones. La sobrevida de estos pacientes es de 2,5 a 3,5 años11 ya que generalmente es progresiva.
El LBA contiene principalmente neutrófilos aunque puede tener leve o moderada elevación de eosinófilos y si exceden el 20% se debe sospechar en enfermedad pulmonar eosinofílica12 ,13.
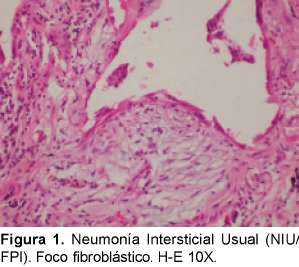
La característica radiológica más prominente es la opacidad reticular periférica que es más marcada en las bases y está asociada a panal de abejas y pérdida de volumen de los lóbulos inferiores. La radiografía simple ocasionalmente puede ser normal. La TACAR muestra opacidades reticulares asociadas con bronquiectasias de tracción y con panal de abejas con atenuación en vidrio esmerilado de predominio basal y periférico. Desde el punto de vista histológico el diagnóstico diferencial es con la asbestosis, drogas o infecciones14, enfermedades de tejido conectivo y con otras NII. El consenso SAT/SER ha recomendado el término de FPI al cuadro clínico de tos crónica y disnea progresiva asociado a una enfermedad intersticial restrictiva y hallazgos histopatológicos de NIU. El diagnóstico definitivo de la FPI requiere de la valoración histopatológica y la exclusión de causas secundarias de EPID como drogas, ETC o exposiciones ambientales (Ver figuras 1 y 2). En la Tabla 5 podemos observar los criterios diagnósticos de la FPI sin necesidad de realizar la biopsia pulmonar.
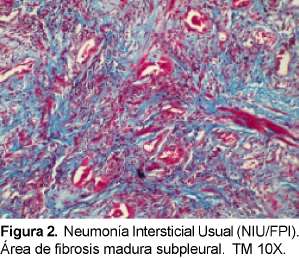

Se han descrito varias ETC asociadas al patrón de NIU como la artritis reumatoide (AR), esclerosis sistémica (ES), polimiositis/dermatomiositis (PM/ DM), síndrome de Sjogren15. Es la entidad que es menos probable que responda a los corticoides16 en comparación con las otras neumonías intersticiales. Puede estar asociada a un peor pronóstico en comparación con las otras neumonías intersticiales aunque no se han comparado los diferentes subtipos de EPI en pacientes con una misma ETC o un subtipo de neumonía intersticial comparándolas en diferentes ETC. Se ha sugerido que la vida media de los pacientes con EPI (definida como alveolitis fibrosante) asociada a AR es de menos de tres años desde el momento del diagnóstico (definido en el momento que fue registrado en la historia) y por lo tanto no es mejor que la vida media de los pacientes con fibrosis pulmonar idiopática17. Sin embargo, en este estudio no se subclasificaron los diferentes subtipos de la EPI. Además, otros estudios aunque limitados han informado el curso más benigno de la EPI asociada a AR en comparación con la FPI18. Muchos de los estudios realizados tratando de aclarar este punto han sido realizados con un número pequeño de pacientes, resultados histopatológicos inconsistentes y un sesgo en la inclusión de pacientes porque se han incluido pacientes con un mal pronóstico y no se incluyeron pacientes con menor afectación pulmonar.
Neumonía intersticial no específica (NINE)
Se decidió dar este nombre a esta entidad con base en su patrón histológico y hasta que se determine la naturaleza definitiva de su entidad clínica. Es sorprendente que luego de los esfuerzos por unificar criterios no se tenga la certeza de que esta entidad sea una entidad separada y específica. Por tanto se considera que este es un término temporal. Sin embargo, la separación y la individualización de las otras entidades, especialmente de la FPI, han ayudado al entendimiento de la NII.
Aunque se considera que la NINE puede tener un patrón fibroso significativo, esta fibrosis es característicamente uniforme en el tiempo a diferencia de la FPI/NIU y el panal de abejas y los focos fibroelásticos son raros (Figuras 3 y 4). En un paciente podemos encontrar patrones histológicos mezclados, en particular la NINE y la NIU; así es que cuando se esté ante este cuadro se debe considerar que el cuadro pronóstico predominante es el de NIU19. Una minoría de pacientes con NINE puede morir. Cuando estamos ante un patrón NINE debemos estar atentos a descartar inicialmente una causa secundaria y además de que puede ser la manifestación inicial de una enfermedad de tejido conectivo. También se considera que puede ser la única manifestación de una neumonitis por hiper-sensibilidad. Varias infecciones pueden presentar este patrón, especialmente el virus de la inmunodeficiencia humana.
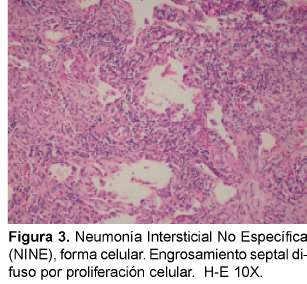
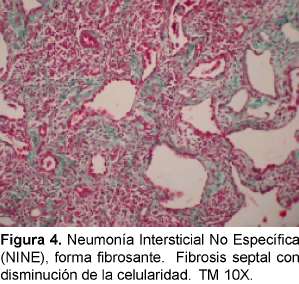
Las características clínicas en los pacientes que presentan el patrón histológico de NINE están poco definidas. Se considera que son un grupo heterogéneo y que puede haber subgrupos. La edad de presentación es cerca de una década más temprano que la FPI y puede verse en niños y suele ser gradual. No se relaciona con género ni con el tabaquismo. Los síntomas usuales son disnea, pérdida de peso (6Kg en promedio) y fatiga con un promedio de duración de 18 a 31 meses. Se puede ver en una minoría fiebre e hipocratismo digital en 10 a 35% de los pacientes. Se ausculta inicial-mente estertores basales que luego se pueden generalizar. En el LBA podemos ver linfocitos en el 50% y en el otro 50% neutrófilos y eosinófilos que con las otras características nos sugieren fuertemente NINE.
Se caracteriza en la radiografía por tener un patrón de infiltrados pulmonares o alveolares en las zonas bajas aunque se han descrito infiltrados en parches20 y patrón intersticial21. En la TACAR observamos predominantemente una primacía de vidrio esmerilado bilateral de predominio subpleural. En la mitad de pacientes se observan opacidades lineares o reticulares y están en relación con bronquiectasias de tracción. Son poco comunes las consolidaciones y el patrón en panal de abejas. Existe cada vez más evidencia de que se diferencia con la NIU en la existencia de panal de abejas aunque la TACAR solo es útil en diferenciarlas hasta en el 70%22. El diagnóstico diferencial de la NINE en la TACAR se debe hacer con la NIU, sarcoidosis y la neumonitis por hipersensibilidad.
Las ETC asociadas a NINE que han sido descritas son ES, PM/DM, AR, LES, Sjogren y la EMTC. Tiene mejor pronóstico que la FPI y mejor respuesta a los corticoesteroides, pero depende del patrón histológico dominante (si es celular: inflamatorio o fibrótico)23, 24. Por esto último y porque en muchas ocasiones las NI complicando las ETC no se pueden diferenciar desde el punto de vista clínico y radiológico, se considera que el establecer la lesión histológica tiene consecuencias terapéuticas y pronósticas importantes. Cuando clínicamente se está con un paciente que no responde a los esteroides, uno podría sospechar que está frente a un patrón de FPI/NIU y que si responde estaría a un patrón diferente a la NINE.
Se ha dicho que en pacientes con EPI asociada a ES el cuadro clínico y radiológico es muy semejante a la FPI/NIU y que su pronóstico es mejor25. Esto se explicaría según algunos autores porque el patrón histológico más frecuente en la ES podría ser el de la NINE26, 27 y no precisamente el de FPI/NIU, pero no hay evidencia de que el pronóstico de la NIU asociada a la ES sea mejor a la NINE asociada a la ES.
Neumonía organizante criptogénica (NOC)
Es una entidad clínico-patológica descrita en 1983 por Davison28. Epler describió esta entidad bajo el término de “bronquiolitis obliterante con neumonía organizante” (del inglés BOOP), haciéndose popular su uso. Pero ahora se prefiere el término NOC porque describe exactamente el cuadro clínico y evita confusiones con otras alteraciones de la vía aérea como la bronquiolitis constrictiva obliterante29.
La característica histológica de la NOC es la apariencia temporal uniforme de una inflamación crónica intersticial con “organización” en los conductos alveolares y en los alvéolos con o sin organización en los bronquiolos (bronquiolitis polipoide obliterans) (Ver Figura 5). Se dice que es mejor el término de NOC cuando es idiopática y si está asociada a alguna patología, denominar neumonía organizante asociada a artritis reumatoide o a neumonía viral. Otras causas secundarias son daño alveolar difuso organizante, organización distal a obstrucción, medicamentos y tóxicos, neumonitis por hipersensibilidad, enfermedad intestinal inflamatoria y síndromes eosinofílicos. Existe un predominio similar en los dos géneros, en los no fumadores es más frecuente que en los fumadores en proporción de 2:1. Existen grados variables de tos y disnea con esputo claro o pálido. Algunas veces el cuadro sigue a una infección respiratoria tipo neumonía adquirida en la comunidad tratada con antibióticos. Son comunes la fiebre intermitente, pérdida de peso, escalofríos, sudoración y mialgias y los estertores difusos sin demostrar consolidación. No se observa hipocratismo digital y son comunes también la elevación de reactantes de fase aguda y la neutrofilia30. En el LBA se ve un aumento en el número total de linfocitos con una relación CD4: CD8 disminuida (aunque al inicio) y se ve aumento de eosinóflos y neutrófilos. Un aumento excesivo de eosinófilos debe alertar de una neumonía eosinofílica.
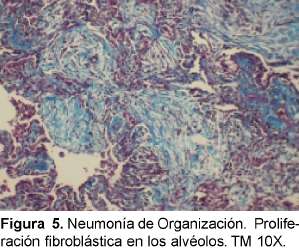
El patrón radiológico es una consolidación unilateral o bilateral31 en parches y se ven lesiones nodulares en 10 a 50% de los casos; una minoría de pacientes pueden mostrar un patrón reticulo-nodular. Las áreas de consolidación de la vía aérea se observan en la TACAR en un 90% de los casos32 con distribución subpleural y peribronquial. Pequeños nódulos de menos de 10 mm se ven a lo largo de los paquetes broncovasculares en más del 50% de los casos. Se observa vidrio esmerilado en 60% de los casos asociado a una consolidación. El patrón en panal de abejas es raro. El diagnóstico diferencial radiológico incluye las ETC: vasculitis y sarcoidosis, el carcinoma de células alveolares, linfoma, e infecciones.
Las ETC asociadas a NOC descritas son AR, LES, ES, PM/DM y Sjogren. Las manifestaciones bron-quiolares son más frecuentemente descritas en AR y Sjogren, pero cualquier ETC puede asociarse a NOC. La NOC asociada a ETC responde generalmente alos corticoides y recaen al retirarlos, por lo que es necesario con frecuencia adicionar un inmuno-supresor.
Neumonía intersticial aguda (NIA)
La neumonía intersticial aguda (NIA) es el término clínico del daño alveolar difuso idiopático. El cuadro clínico se inicia con un pródromo similar a una enfermedad viral dado por mialgias, artralgias, fiebre, escalofrío y malestar general. La disnea es común y se auscultan estertores o signos de consolidación. Esta neumonía intersticial se considera que es de curso agudo a diferencia de las otras NII. Las características clínicas están basadas en un amplio rango de edad con una edad media de 50 años y no tiene relación con el tabaquismo. Usualmente existe una progresión de la falla respiratoria que requiere ventilación mecánica y el desarrollo del síndrome de dificultad respiratoria aguda.
Desde el punto de vista histopatológico se describe como una forma organizante del daño alveolar difuso que es indistinguible de la forma histológica del síndrome de dificultad respiratoria adulta (del inglés ARDS) causado por el choque y la sepsis que tiene una distribución difusa con apariencia temporal uniforme. La fase exudativa incluye edema, membranas hialinas e inflamación intersticial aguda (Ver Figura 6). La fase organizante incluye una fibrosis organizante no densa en los septos alveolares e hiperplasia de los neumocitos tipo II (Ver Figura 7). Si el paciente sobrevive, los pulmones restauran parcialmente su arquitectura; en otros casos los pulmones tienen una fibrosis extensa33 (Ver Figura 8).
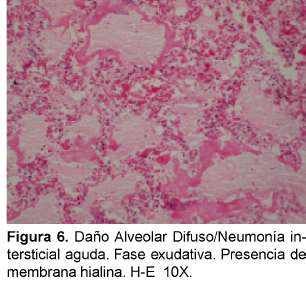
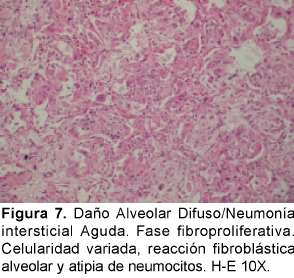
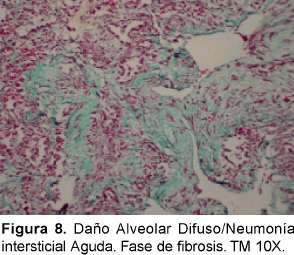
En el LBA se observa un aumento de las células totales, de los glóbulos rojos y la hemosiderina, de neutrófilos y ocasionalmente de linfocitos. Se pueden ver neumocitos atípicos y fragmentos de membranas hialinas. Las radiografías demuestran opacificación bilateral en parches del espacio aéreo, con broncograma aéreo.
Los volúmenes pulmonares están generalmente diminuidos, pero se pueden ver normales. A medida que la enfermedad progresa se observa un patrón de vidrio esmerilado.
En la TACAR el patrón más frecuente es el de vidrio esmerilado que en la fase inicial exudativa es en parches y en forma bilateral de predominio basal con áreas localizadas de parénquima normal, lo que le da la apariencia geográfica34. Estas características la diferencian con el ARDS. Se observan también la dilatación bronquial y la distorsión de la arquitectura. La consolidación basal se ve en la mayoría de casos.
Las ETC asociadas a NIA que han sido descritas son: AR, LES, ES, PM/DM, Sjogren.
Bronquiolitis respiratoria asociada a enfermedad pulmonar intersticial (BR-EPI)
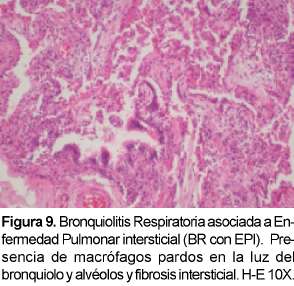
La bronquiolitis respiratoria es una lesión histopatológica que se encuentra en los fumadores de cigarrillo y que en raras ocasiones se presenta asociada a una enfermedad intersticial (Ver Figura 9). Se le ha relacionado con la neumonía descamativa intersticial y por tanto se debe hacer el diagnóstico diferencial con esta entidad. Los síntomas son leves e incluyen disnea y tos pero puede observarse disnea severa o hipoxemia y no suele observarse hipocratismo. Afecta usualmente a fumadores activos en los treinta a cuarenta años con más de 30 paquetes al año. Cuando la BR-EPI se torna sintomática, las pruebas de función pulmonar y las imágenes se tornan positivas35. Las PFP demuestran reducción en DLCO con patrón de restricción y obstrucción de la vía aérea. El LBA contiene macrófagos con inclusiones pigmentadas oro, café o negras e indistinguibles de aquellas de fumadores normales. La característica radiológica más común es el engrosamiento de la pared de los bronquios periféricos o centrales36. La apariencia de vidrio esmerilado se observa en el 60% de los pacientes y se puede ver una radiografía normal hasta en el 14% de ellos. Los hallazgos en la TACAR incluyen nódulos centrilobulares, atenuación en vidrio esmerilado y engrosamiento de vías aéreas centrales y periféricas que se puede encontrar en fumadores pero que usualmente en esta patología es más severo. Es común el enfisema centrilobular leve en los lóbulos superiores con áreas de hipoatenuación por atrapamiento aéreo. El diagnóstico diferencial en la TACAR incluye neumonitis por hi-persensibilidad aguda, la NDI y la NINE.
Neumonía descamativa intersticial (NDI)
Este término se usó originalmente porque se creyó que el cuadro histológico predominante era de descamación de células epiteliales, pero se sabe que es la acumulación intra-alveolar de macrófagos37 (ver Figura 10). Esta entidad se considera por muchos autores como el final del espectro clínico de BR-EPI debido a su similitud en la histopatología y porque se asocia a fumar cigarrillo38 ,39. Afecta a fumadores en la cuarta o quinta década de vida. Predomina más en hombres que en mujeres con síntomas de disnea y tos, puede llegar a falla respiratoria y se puede ver hipocratismo digital en la mitad de los pacientes40. La radiografía de tórax puede ser normal entre el 3 y el 22% de los pacientes41. Puede observarse un patrón generalizado de parches en vidrio esmerilado con predominio en lóbulos inferiores42. En la TACAR se observa opacificación en vidrio esmerilado en todos los casos de NDI. Se observan opacidades lineares irregulares y un patrón reticular en el 59% de pacientes y panal de abejas en menos de la tercera parte43.Tiene además predominio de las zonas inferiores en la mayoría de los casos. El diagnóstico diferencial radiológico incluye la neumonitis por hipersensibilidad aguda o subaguda, la sarcoidosis e infecciones tales como la del pneumocistis carinii. El pronóstico es bueno y los pacientes mejoran con corticoides y con dejar de fumar.
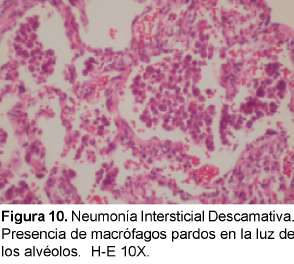
Se han descrito casos de NDI en LES, AR y ES en muy pocos casos. No ha sido claro que los casos descritos hayan sido por el tabaquismo o la ETC subyacente3.
Neumonía intersticial linfoide (NIL)
Su incidencia es baja. Su existencia es dudosa para algunos autores mientras que otros dicen que se debe excluir de la clasificación de las neumonías intersticiales y se debe incluir en las hiperplasias linfoides de los desórdenes linfoproliferativos pulmonares. La presentación clínica es poco definida. Se presenta más en mujeres y luego de la quinta década de vida44. Suele presentarse tos y disnea gradual y en ocasiones fiebre, dolor torácico, pérdida de peso y artralgias. Al examen físico se pueden encontrar estertores y adenopatía, esto último más frecuentemente asociado a síndrome de Sjögren45.
Se puede ver asociación con otras ETC e inmunodeficiencias. Se puede observar anemia y una elevación policlonal o monoclonal de las inmunoglobulinas causando esto sospecha de un desorden mieloproliferativo. Se han descrito dos patrones radiológicos, uno basilar con componente alveolar o el otro difuso con patrón en panal de abejas46. El hallazgo en la TACAR es generalmente en vidrio esmerilado y en cerca de la mitad de los pacientes se puede ver un patrón reticular y se pueden ver nódulos y una consolidación difusa. También se han descrito nódulos centrolobulares y subpleurales y un en-grosamiento de los septos interlobulares y de los paquetes broncovasculares47.
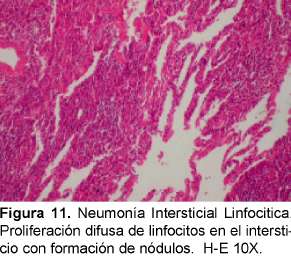
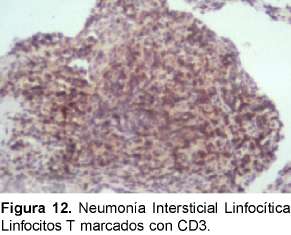
Se debe diferenciar histológicamente con la bronquiolitis folicular que se caracteriza por un infiltrado linfocítico peribronquial con centros germinales (Ver Figuras 11 y 12). La diferenciación desde el punto de vista radiológico es con la neumonitis por hipersensibilidad, sarcoidosis y la diseminación linfangítica de tumores. Se ha descrito esta entidad asociada, además de Sjogren, a LES, AR, EMTC y a miopatías inflamatorias.
Proceso diagnóstico
El diagnóstico de las NII es un proceso dinámico que incluye la parte clínica, la radiología y la patología. No existe un solo estándar de oro para su diagnóstico. La evaluación clínica incluye la realización de pruebas de función pulmonar que demuestren patrón restrictivo principalmente48. Es de anotar que se requiere la técnica establecida para la TACAR49 y que es importante la experiencia del radiólogo para el diagnóstico de NII y su distinción con otras EPPD50. La TACAR ayuda en la evaluación de la NII cuando existe una colaboración entre el clínico y el radiólogo51. La TACAR ha demostrado eficacia en EPI en identificar la presencia de la enfermedad (es anormal muchas veces cuando otros parámetros como pruebas funcionales son normales), en la evaluación de la extensión de la enfermedad, en definir el sitio de la biopsia y evaluar la evolución de la enfermedad, en el tratamiento y el diagnóstico diferencial. Igualmente la exactitud diagnóstica de la TACAR no se ha estudiado en estudios poblacionales no seleccionados. Por tanto es importante hacer énfasis en la necesidad de realizar biopsia pulmonar para estudiar esta patología en sus aspectos de historia natural, identificación de subtipos, pronóstico, respuesta al tratamiento y como diagnóstico diferencial con otras patologías diferentes a las NII. Desafortunadamente, por la falta de especificidad diagnóstica de la TACAR, no se ha reemplazado la necesidad de realizar la biopsia pulmonar en muchos de los casos para llegar a un diagnóstico definitivo52.
Los criterios histopatológicos para el diagnóstico de las NII se han aceptado cada vez, pero existe todavía poca correlación intra-observador incluso entre los patólogos expertos53. La biopsia pulmonar ha demostrado tener una gran rendimiento diagnóstico (92%) y una incidencia baja en la morbilidad (2,5%) y la mortalidad (0,2%) y con la introducción de la video-laparoscopia se ha mejorado la técnica de la biopsia54. Por tanto se considera que el no apoyarse en la biopsia es injustificado debido a que algunos estudios han demostrado que incluso expertos en EPI pueden realizar un diagnóstico confiable solo en el 50% de los casos especialmente cuando el cuadro inicial no corresponde al de FPI/NIU55. El número de muestras a tomar debe ser de más de un lóbulo del pulmón; el sitio de la muestra es tomar del borde de la lesión y que contenga un borde normal y tomada lo más profundo que se pueda de la superficie subpleural. El uso de la biopsia transbronquial para el diagnóstico de las EPID no se recomienda. La utilidad del LBA en las NII tanto para diagnóstico como para evolución y pronóstico ha sido limitada56. Entonces, el estándar de oro para el diagnóstico de las neumonías intersticiales idiopáticas es la combinación de las características clínicas, radiológicas e histopatológicas.
Conclusión
Varias enfermedades pulmonares están asociadas a las ETC. Las NII son parte de este grupo y hay varias entidades a tener en cuenta para su diagnóstico diferencial. Nuevos conceptos se han originado en cuanto a las neumonías intersticiales idiopáticas y por tanto, a las asociadas a ETC. A pesar de su mejor conocimiento en los últimos años, no se ha podido establecer la verdadera naturaleza y frecuencia debido a diferentes criterios para su valoración. No se conoce la frecuencia de EPI en ETC y los factores de riesgo para desarrollarla y no se conocen con exactitud los factores fisiopatológicos para el desarrollo de EPI ni su historia natural. El proceso diagnóstico de las NII requiere de un proceso dinámico clínico, radiológico y patológico y de la colaboración en equipo del clínico, del radiológo y del patólogo. El estándar de oro para el diagnóstico de las neumonías intersticiales idiopáticas es la combinación de las características clínicas, radiológicas e histopatológicas. La biopsia pulmonar es parte fundamental en este proceso Por tanto se debe impulsar entre los reumatólogos una mayor documentación del daño intersticial pulmonar en los pacientes que sufren de enfermedades reumatológicas con el fin de determinar el tipo de lesión, establecer la historia natural, refinar el diagnóstico diferencial y conducir un tratamiento bien dirigido de acuerdo al subtipo de NII con el fin de mejorar su pronóstico.
Referencias
1. Cushley MJ, Davison AG, du Bois RM. The diagnosis, assessment and treatment of diffuse parenchymal lung disease in adults. Thorax 1999; 54: S1-S30. [ Links ]
2. Travis, A William D, King T E. American Thoracic Society: Idiopathic pulmonary fibrosis: diagnosis and treatment. International Consensus Statement. American Thoracic Society Lung fibrosis: classification and therapy Veerara-ghavan et al.503 (ATS) and the European Respiratory Society (ERS). Am J Respir Crit Care Med 2000; 161: 646-664. [ Links ]
3. Vassallo R, Thomas Ch. Advances in the treatment of rheumatic interstitial lung disease. Curr Opin Rheumatol 2004; 16: 186-191. [ Links ]
4. Bjoraker JA, Ryu JH, Edwin MK, et al. Prognostic significance of histopathologic subsets in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 1998; 157: 199-203. [ Links ]
5. Ward MM, Donald F. Pneumocystis carinii pneumonia in patients with connective tissue diseases: the role of hospital experience in diagnosis and mortality. Arthritis Rheum 1999; 42: 780-789. [ Links ]
6. Kadoya A, Okada J, Iikuni Y. Risk factors for Pneumocystis carinii pneumonia in patients with polymyositis/derma-tomyositis or systemic lupus erythematosus. J Rheumatol 1996; 23: 1186-1188. [ Links ]
7. Bachelez H, Schremmer B, Cadranel J. Fulminant Pneumocystis carinii pneumonia in 4 patients with dermatomyositis. Arch Intern Med 1997; 157: 1501-1503. [ Links ]
8. Houtman PM, Stenger AA, Bruyn GA, Methotrexate may affect certain T lymphocyte subsets in rheumatoid arthritis resulting in susceptibility to Pneumocystis carinii infection. J Rheumatol 1994; 21: 1168-1170. [ Links ]
9. Tai TL, O’Rourke KP, McWeeney M. Pneumocystis carinii pneumonia following a second infusion of infliximab. Rheumatology (Oxford) 2002; 41: 951-952. [ Links ]
10. Demedts M, Wells AU, Anto JM. Interstitial lung diseases: an epidemiological overview. Eur Respir J 2001; 18(Suppl 32): 2S-16S. [ Links ]
11. King TE Jr, Costabel U, Cordier J-F, doPico GA, du Bois RM, Lynch D, Lynch JP III, et al.Idiopathic pulmonary fibrosis: diagnosis and treatment. Am J Respir Crit Care Med 2000; 161: 646-664. [ Links ]
12. Anonymous. Bronchoalveolar lavage constituents in healthy individuals, idiopathic pulmonary fibrosis, and selected comparison groups. The BAL Cooperative Group Steering Committee. Am Rev Respir Dis 1990; 141: S169-S202. [ Links ]
13. Allen JN, Davis WB. Eosinophilic lung diseases. Am J Respir Crit Care Med 1994; 150: 1423-1438. [ Links ]
14. Katzenstein AL, Fiorelli RF. Nonspecific interstitial pneumonia/fibrosis. Histologic features and clinical significance. Am J Surg Pathol 1994; 18: 136-147. [ Links ]
15. Kim E, Lee K, Johkoh T, Kim T, Suh G, Kwon J, Han J. Interstitial Lung Diseases Associated with Collagen Vascular Diseases: Radiologic and Histopathologic Findings. Radio-graphics 2002; 22(S150): 1-15. [ Links ]
16. Ryu, J, Bongartz, T, Matteson E. Interstitial Lung disease in connective Tissue Diseaases: What Are the Important Questions?. Arthristis and Rheumatism 2005; 53(4): 488-490. [ Links ]
17. Hubbard R, Venn A. The impact of coexisting connective tissue disease on survival in patients with fibrosing alveolitis. Rheumatology (Oxford) 2002; 41: 676-679. [ Links ]
18. Saravanan V, Kelly CA: Survival in fibrosing alveolitis associated with rheumatoid arthritis is better than cryptogenic fibrosing alveolitis. Rheumatology (Oxford) 2003; 42: 603-605. [ Links ]
19. Flaherty KR, Travis WD, Colby TV, Toews GB, Kazerooni EA, Gross BH, Jain A, Strawderman RL, Flint A, Lynch JP, et al. Histopathologic variability in usual and nonspecific interstitial pneumonias. Am J Respir Crit Care Med 2001; 164: 1722-1727. [ Links ]
20. Park JS, Lee KS, Kim JS, Park CS, Suh YL, Choi DL, Kim KJ. Nonspecific interstitial pneumonia with fibrosis: radiographic and CT findings in seven patients. Radiology 1995; 195: 645-648. [ Links ]
21. Katzenstein AL, Fiorelli RF. Nonspecific interstitial pneumonia/fibrosis. Histologic features and clinical significance. Am J Surg Pathol 1994; 18: 136-147. [ Links ]
22. Elliott TL, Lynch DA, Newell JD Jr, Cool C, Tuder R, Markopoulou K, et al. High-resolution computed tomography features of nonspecific interstitial pneumonia and usual interstitial pneumonia. J Comput Assist Tomogr 2005; 29: 339-345. [ Links ]
23. Katzenstein AL, Fiorelli RF. Nonspecific interstitial pneumonia/fibrosis. Histologic features and clinical significance. Am J Surg Pathol 1994; 18: 136-147. [ Links ]
24. Cottin V, Donsbeck AV, Revel D, Loire R, Cordier JF. Nonspecific interstitial pneumonia. Individualization of a clinicopathologic entity in a series of 12 patients. Am J Respir Crit Care Med 1998; 158: 1286-1293. [ Links ]
25. White B. Interstitial lung disease in scleroderma. Rheum Dis Clin North Am 2003; 29: 371-390. [ Links ]
26. Bouros D, Wells AU, Nicholson AG, et al. Histopathologic subsets of fibrosing alveolitis in patients with systemic sclerosis and their relationship to outcome. Am J Respir Crit Care Med 2002; 165: 1581-1586. [ Links ]
27. Kim DS, Yoo B, Lee JS, et al. The major histopathologic pattern of pulmonary fibrosis in scleroderma is nonspecific interstitial pneumonia. Sarcoidosis Vasc Diffuse Lung Dis 2002; 19: 121-127. [ Links ]
28. Davison AG, Heard BE, McAllister WAC, Turner-Warwick ME. Cryptogenic organizing pneumonitis. Q J Med 1983; 207: 382-394. [ Links ]
29. Travis A, William D, King TE. American Thoracic Society: Idiopathic pulmonary fibrosis: diagnosis and treatment. International Consensus Statement. American Thoracic Society Lung fibrosis: classification and therapy Veerara-ghavan et al.503 (ATS) and the European Respiratory Society (ERS). Am J Respir Crit Care Med 2000; 161: 646-664. [ Links ]
30. King TE Jr, Mortenson RL. Cryptogenic organizing pneumonitis. The North American experience. Chest 1992; 102: 8S-13S. [ Links ]
31. Müller NL, Guerry-Force ML, Staples CA, Wright JL, Wiggs B, Coppin C, Paré P, Hogg JC. Differential diagnosis of bronchiolitis obliterans with organizing pneumonia and usual interstitial pneumonia: clinical, functional, and radiologic findings. Radiology 1987; 162: 151-156. [ Links ]
32. Lee KS, Kullnig P, Hartman TE, Muller NL. Cryptogenic organizing pneumonia: CT findings in 43 patients. Am J Roentgenol 1994; 162: 543-546. [ Links ]
33. Katzenstein AL, Myers JL, Mazur MT. Acute interstitial pneumonia. A clinicopatologic, ultraestructural, and cell kinetic study. Am J Surg Pathol 1986; 10: 256-267. [ Links ]
34. Primack SL, Hartman TE, Ikezoe J, Akira M, Sakatani M, Muller NL. Acute interstitial pneumonia: radiographic and CT findings in nine patients. Radiology 1993; 188: 817-820. [ Links ]
35. Wittram, C. The idiopathic interstitial pneumonias. Curr Probl Diagn Radiol 2004; 33: 189-199. [ Links ]
36. Heyneman LE, Ward S, Lynch DA, Remy-Jardin M, Johkoh T, Muller NL. Respiratory bronchiolitis, respiratory bronchiolitis-associated interstitial lung disease, and desquamative interstitial pneumonia: different entities or part of the spectrum of the same disease process? Am J Roentgenol 1999; 173: 1617-1622. [ Links ]
37. Liebow AA, Steer A, Billingsley JG. Desquamative interstitial pneumonia. Am J Med 1965; 39: 369-404. [ Links ]
38. Myers JL, Veal CF Jr, Shin MS, Katzenstein AL. Respiratory bronchiolitis causing interstitial lung disease. A clinicopa-thologic study of six cases. Am Rev Respir Dis 1987; 135: 880-884. [ Links ]
39. Niewoehner DE, Kleinerman J, Rice DB. Pathologic changes in the peripheral airways of young cigarette smokers. N Engl J Med 1974; 291: 755-758. [ Links ]
40. Carrington CB, Gaensler EA, Coutu RE, FitzGerald MX, Gupta RG. Natural history and treated course of usual and desquamative interstitial pneumonia. N Engl J Med 1978; 298: 801-809. [ Links ]
41. Gaensler EA, Goff AM, Prowse CM. Desquamative interstitial pneumonia. N Engl J Med 1966; 274: 113-128. [ Links ]
42. Feigin DS, Friedman PJ. Chest radiography in desquamative interstitial pneumonitis: a review of 37 patients. Am J Roentgenol 1980; 134: 91-99. [ Links ]
43. Hartman TE, Primack SL, Swensen SJ, Hansell D, McGuinness G, Muller NL. Desquamative interstitial pneumonia: thin-section CT findings in 22 patients. Radiology 1993; 187: 787-790. [ Links ]
44. Strimlan CV, Rosenow EC, Weiland LH, Brown LR. Lymphocytic interstitial pneumonitis. Review of 13 cases. Ann Intern Med 1978; 88: 616-621. [ Links ]
45. Liebow AA, Carrington CB. Diffuse pulmonary lymphore-ticular infiltrations associated with dysproteinemia. Med Clin North Am 1973; 57: 809-843. [ Links ]
46. Julsrud PR, Brown LR, Li CY, Rosenow EC, Crowe JK. Pulmonary processes of mature-appearing lymphocytes: pseudolymphoma, welldifferentiated lymphocytic lymphoma, and lymphocytic interstitial pneumonitis. Radiology 1978; 127: 289-296. [ Links ]
47. Johkoh T, Muller NL, Pickford HA, et al. Lymphocytic interstitial pneumonia: thin section CT findings in 22 patients. Radiology 1999; 212: 567-572. [ Links ]
48. Roland M. du Bois. Evolving Concepts in the Early and Accurate Diagnosis of Idiopathic Pulmonary Fibrosis. Clin Chest Med 2006; 27: S17-S25. [ Links ]
49. Austin JH, Muller NL, Friedman PJ, Hansell DM, Naidich DP, Remy- Jardin M, Webb WR, Zerhouni EA. Glossary of terms for CT of the lungs: recommendations of the Nomenclature Committee of the Fleischner Society. Radiology 1996; 200: 327-331. [ Links ]
50. Lynch DA, Godwin JD, Safrin S, Starko KM, Hormel P, Brown KK, Raghu G, King TE Jr, Bradford WZ, Schwartz DA, et al. Idiopathic Pulmonary Fibrosis Study Group. High-resolution computed tomography in idiopathic pulmonary fibrosis: diagnosis and prognosis. Am J Respir Crit Care Med 2005; 172: 488-493. [ Links ]
51. Flaherty KR, King TE Jr, Raghu G, Lynch JP III, Colby T V, Travis WD, et al. Idiopathic interstitial pneumonia: what is the effect of a multidisciplinary approach to diagnosis? Am J Respir Crit Care Med 2004; 170: 904-910. [ Links ]
52. Andersen HA. Transbronchoscopic lung biopsy for diffuse pulmonary diseases: results in 939 patients. Chest 1978; 73: 734-736. [ Links ]
53. Nicholson AG, Addis BJ, Bharucha H, Clelland CA, Corrin B, Gibas AR, Hasleton PS, Kerr KM, Ibrahim NB, Stewart S, et al. Interobserver variation between pathologists in diffuse parenchymal lung disease. Thorax 2004; 59: 500-505. [ Links ]
54. King TE Jr. Clinical advances in the diagnosis and therapy of the interstitial lung diseases. Am J Respir Crit Care Med 2005; 172: 268-279. [ Links ]
55. Flaherty KR, King TE Jr, Raghu G, Lynch JP III, Colby T V, Travis WD, et al. Idiopathic interstitial pneumonia: what is the effect of a multidisciplinary approach to diagnosis?. Am J Respir Crit Care Med 2004; 170: 904-910. [ Links ]
56. Reynolds HY. Use of bronchoalveolar lavage in humans: past necessity and future imperative. Lung 2000; 178: 271-293. [ Links ]

