










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Reumatología
Print version ISSN 0121-8123
Rev.Colomb.Reumatol. vol.35 no.3 Bogotá July/Sept. 2007
Artículo de Revisión
Small vessel vasculitis
History, classification, etiology, histopathology, clinic, diagnosis and treatment
Vasculitis de pequeños vasos
Historia, clasificación, etiología, histopatología, clínica, diagnóstico y tratamiento
Antonio Iglesias Gamarra1, Eric L. Matteson2, José Félix Restrepo3
1 Profesor Titular de Medicina Interna y Reumatología. Universidad Nacional de Colombia.
2 Division of Rheumatology, Mayo Clinic College of Medicine.
3 Profesor Titular de Medicina Interna y Reumatología. Universidad Nacional de Colombia.
Recibido: junio 29/2007 Aceptado: agosto 30/2007
Summary
Small-vessel vasculitis is a convenient descriptor for a wide range of diseases characterized by vascular inflammation of the venules, capillaries, and/or arterioles with pleomorphic clinical manifestations. The classical clinical phenotype is leukocytoclastic vasculitis with palpable purpura, but manifestations vary widely depending upon the organs involved. Histopathologic examination in leukocytoclastic vasculitis reveals angiocentric segmental inflammation, fibrinoid necrosis, and a neutrophilic infiltrate around the blood vessel walls with erythrocyte extravasation. The etiology of small-vessel vasculitis is unknown in many cases, but in others, drugs, post viral syndromes, malignancy, primary vasculitis such as microscopic polyarteritis, and connective tissue disorders are associated. The diagnosis of small-vessel vasculitis relies on a thorough history and physical examination, as well as relevant antibody testing including antinuclear antibody and antineutrophil cytoplasmic antibody, hepatitis B and C serologies, assessment of complement, immunoglobulins, blood count, serum creatinine, liver function tests, urinalysis, radiographic imaging, and biopsy. The treatment is based primarily on corticosteroid and immunosuppressive agents.
Key words: vasculitis, small vessel vasculitis, leukocytoclastic vasculitis, linphomonocitic vasculitis, ANCA associated vasculitis.
Resumen
El término vasculitis de pequeños vasos describe a un grupo de enfermedades caracterizadas por inflamación de vénulas, capilares y/o arteriolas con manifestaciones clínicas pleomórficas. El fenotipo clínico clásico es la vasculitis leucocitoclástica con púrpura palpable, pero con manifestaciones que varían ampliamente dependiendo del órgano comprometido. La histología en la vasculitis leucocitoclástica revela una inflamación segmentaria angiocéntrica, necrosis fibrinoide e infiltrado neutrofílico alrededor de los vasos sanguíneos, con extravasación de eritrocitos. La etiología de las vasculitis de pequeños vasos es desconocida, en muchos casos, pero en otros se ha asociado con drogas, síndromes post virales, neoplasias, vasculitis primarias como la poliarteritis microscópica, y enfermedades del tejido conjuntivo. El diagnóstico de las vasculitis de pequeños vasos se basa en la historia clínica y el examen físico, así como con estudio de anticuerpos como los anticuerpos antinucleares y los anticuerpos contra el citoplasma de los neutrófilos, serología de hepatitis B y C, determinación de inmunoglobulinas, complemento, creatinina sérica, función renal, urianálisis, estudios de imágenes y biopsia. El tratamiento se basa primariamente en el uso de corticosteroides e inmunosupresores.
Palabras clave: vasculitis, vasculitis de pequeños vasos, vasculitis leucocitoclástica, vasculitis linfomonocítica, vasculitis asociada a ANCA.
Introduction
The systemic inflammatory vascular diseases are a heterogeneous group of conditions whose common feature is that of vessel inflammation. This vasculitis is characterized by fibrinoid necrosis, thrombosis, and sometimes a granulomatous reaction1 . Vascular damage may occur in venules, capillaries, and arterioles, causing local and systemic clinical manifestations, depending on the organs involved. Vessels of any type in any organ can be affected, a fact that result in a wide variety of sign and symptoms. The clinical picture of small vessel vasculitis is also dependent on the extent of vascular bed involvement, delay in diagnosis, and treatment1. These heterogeneous clinical manifestations, combined with the etiologic non specificity of the histologic lesions, complicate the diagnosis of specific form of vasculitis.
Recognition of these features of vasculitis and evaluation with selected laboratory and other clinical tests and histologic evaluation of biopsy specimens generally permits a specific diagnosis, which directs the evaluation of activity, extent, and damage, and guides treatment. However, signs and symptoms of various forms of vasculitis are overlapping, and diagnostic precision is often hampered by the lack of diagnosis-specific histologic findings. This creates a clinical dilemma, because the treatment and prognosis of specific forms of vasculitis varies. For example, a patient with cutaneous leukocytoclastic vasculitis with abdominal and renal involvement may have disease due to classic polyarteritis nodosa (PAN) or to microscopic polyangiitis, or Henoch-Schönlein purpura. Indeed, there are few clinical conditions which cause as much confusion and consternation among clinicians and patients alike as do the protean presentations and management of vasculitis1,2.
The gold standard for a diagnosis of vasculitis is histologic confirmation on biopsy, as few forms of vasculitis have a pathognomonic laboratory or imaging finding. Interpretation of the biopsy sample is dependent on a number of variables, including the interest and experience of the pathologist, tissue selection and quantity, and the amount of time which has occurred between diseases onset and obtaining the sample. These variables affect and aid verification of the diagnosis. A positive biopsy supports the diagnosis, while a negative one does not necessarily exclude it. This may be the case, for example, when vasculitis affects an organ or appendage which is poorly amenable to biopsy or biopsy of apparently involved tissue demonstrates a non-inflammatory vasculopathy1,2.
Small vessel vasculitis with cutaneous involvement does not constitute a subgroup of either primary or secondary vasculitis. Rather, it can be associated with a number of comorbidities, further complicating diagnosis and, hence, treatment decisions. As an example, patients with small vessel disease may have hepatitis B or C, with or without cryoglobulinemia.
The discovery that autoantibodies against cytoplasmic antigens of neutrophils (anti-neutrophil cytoplasmic antibodies (ANCA)) are closely associated with vasculitic disorders has improved diagnosis of patients with clinically suspected vasculitis and/or glomerulonephritis (GN). Like the introduction of ANA serology for systemic lupus erytematosus, introduction of ANCA testing for vasculitis has revealed myriad clinicopathological presentations beyond the previously recognized patterns of systemic disease3. As a clinicopathological process, vasculitis occurs both as a primary process or idiopathic vasculitis and as a secondary feature of other diseases secondary vasculitis such as collagen vascular diseases, infectious disorders, malignancy and adverse drugs reaction.
The vasculitic syndromes share a common histopathological substrate inflammation within blood vessels resulting in vascular obstruction with tissue ischemia and infarction. Focal necrotizing lesions are the common vascular pathology that characterizes the ANCA-associated disorders: Wegener's granulomatosis (WG), microscopic polyangiitis (MPA) and Churg-Strauss syndrome (CSS). These lesions can affect many types of vessel and lead to a variety of symptoms and signs. Immunohistology shows little deposition of immune reactants, a feature which distinguishes lesions due to ANCA-associated vasculitis (AAV) from those of antiglomerular basement membrane disease, IgA nephropathy, and lupus nephritis3.
ANCA were first described in 1982 by Davies and his associates as a cause of diffuse granular cytoplasmic inmuno-fluorescence staining (C-ANCA) on ethanol-fixed neutrophils in association with glomerulonephritis, vasculitis and Wegener's granulomatosis4. Two years later, Hall et al. confirmed this observation in four patients with small-vessel vasculitis5. Van der Woude et al.6 in 1985 generated substantial interest by suggesting that detection of ANCA was a useful diagnostic and prognostic marker for Wegener's granulomatosis. Subsequent work by Van der Woude et al. Falk et al. and others demonstrated that ANCA are closely associated with three major categories of small-vessel vasculitis: Wegener's granulomatosis, microscopic polyangiitis, and Churg-Strauss syndrome6-15. These forms of vasculitis have subsequently been grouped together and are referred to as ANCA associated vasculitis14.
The basis for the appearance of ANCA is not understood. One favored hypothesis is that environmental factors such infectious pathogen is required to activate the pre-existing potential autoimmune cellular repertoire. The proposed mechanisms by which infections break self tolerance can include bystander damage, unveiling of hidden self epitope, molecular mimicry and determinant molecular spreading. There is evidence that certain types of environmental exposure, to silica, for example, or to infectious pathogens, are associated with AAV3, 16-19. These mechanisms likely play a role in other forms of vasculitis as well.
Historical background
The first historical accounts of vasculitis are of small vessel vasculitis, especially forms associated with purpura. The Latin term vasculitis may have derived from the Greek porphyra, describing the color produced by a mollusk (purpura lapillus)20. By the XVI century, the word purpura had begun to refer to infectious diseases with fever, such as typhoid fever, but was also being used to describe other conditions often referred to as "purpura sine fever", "petechia sine fever," and the term palpable purpura carne into use20. The English dermatologist Willan classified purpura as simple, hemorrhagic, urticarial, and contagious20, 21. The concept of hemorrhagic fever he introduced in 1808 was expanded on by Bauer in 182421, 22. A unifying concept of purpura and its relationship to leukocytoclastic vasculitis was put forward by Zeek et al. in 1948 and 1952, who called this form of vasculitis with small vessel involvement hypersensitivity angitis23, 24. Davson et al. and Godman et al. referred to it as microscopic polyangiitis, a concept adopted by the Chapel Hill international consensus conference in 1994, at which time several forms of small vessel vasculitis were more clearly defined15, 25, 26.
This progress was the direct result of William's early work. He clearly distinguished purpura caused by systemic febrile infections from non infectious purpura20, 21, 27. Drawing on the earlier work of Riverius and Werlhof, Willan assigned to the ancient term purpura the meaning it retains to this day20, 21, 27. He considered the condition at length in this masterwork on cutaneous diseases (1808), and cases of palpable purpura consistent with Henoch-Schônlein syndrome can be recognized in both the text and the plates of his book27. Willan noted that non infectious purpura had a predilection for the lower extremities, was characterized by recurrent groups of lesions, and could be associated with different systemic disease. Schonlein, Henoch, and later Osler and others elucidated a broad spectrum of signs and symptoms that were associated with purpura and small-vessel vasculitis, including arthritis, peripheral neuropathy, abdominal pain, pulmonary hemorrhage, epistaxis and nephritis28-32. Osler recognized that these clinical manifestations were caused by necrotizing inflammation in small vessels26, 27. Other early descriptions were provided by Heberden, the describer of rheumatoid arthritis, in 1801 in his work "Commentarii de morborum historia et curatione"33.
In October, 1893, Amy, age 6 years, was admitted to the Victoria Hospital for Sick children (London). She exhibited firm, tender, sharply defined, pale purplish-red nodules on the hands, elbows, knees, and buttocks. The patient was examined by Henry Radcliffe-Crocker (1845-1909), who was England's Mr. Dermatology at the time, as well as Jonathan Hutchinson (1828-1913), who pointed out the similarity to a case reported earlier by Judson Bury27. Radcliffe-Crocker named the condition erythema elevatum diutinum34. He failed, however, to identify the condition as a form of vasculitis, perhaps because the lesions chosen for biopsy were too mature. Characteristic leukocytoclasia is ordinary prominent only in the early stages of the disease27, 34. In 1929, Fred Weidman and John Besancon of the University of Pennsylvania described the vasculitis of this condition35. This disease is considered to be a strictly cutaneous leukocytoclastic vasculitis, and is not mentioned in the classic classifications of vasculitis, although we consider that it likely represents a primary form of vasculitis, at least in its initial stages...
The classic description of vasculitis is that of Adolf Kussmaul and Rudolf Maier in 1866. They reported on a 27 year old patient who suffered a fulminant disease characterized by fever, productive cough, malaise, weight loss, myalgias, paresthesias and polyneuropathy, proteinuria and abdominal pain. They called the condition periarteritis nodosa, which later evolved into the more pathologically correct name polyarteritis nodosa20-36. For more than 50 years thereafter, and unfortunately even today in some settings, any patient with necrotizing arteritis was given a diagnosis of polyarteritis nodosa.
The first description of microscopic polyangiitis was by Friedrich Wohlwill in Germany in 192337-40. Wohlwill effectively distinguished microscopic polyangiitis from polyarteritis nodosa, and Davson used the presence or absence of glomerulonephritis to separate classic polyarteritis nodosa, in which glomerulonephritis is absent, from polyarteritis nodosa24, an observation adopted in the Chapel Hill vasculitis nomenclature25. The Chapel Hill authors preferred the term "microscopic polyangiitis: to "microscopic polyarteritis" to more accurately describe the small vessel involvement of arterials, venules, and capillaries25, 26.
Another form of vasculitis associated with but not confined to small vessel inflammation is Wegener's granulomatosis, a systemic inflammatory disease with a broad clinical spectrum. The disease was first described in 1931 by a medical student at the Charité in Berlin, Heinz Klinger, who mistakenly believed it to be an atypical form of polyarteritis nodosa41, 42.
At the 29th Meeting of the German Society of Pathology in Breslau, Friedrich Wegener, a good friend of Klinger's, reported on the post-mortem findings in three of his patients41. He reported 11 patients in detail in 1939 as an assistant at the Pathology Institute of the University of Breslau27, 43. Initial symptoms included sniffles and progressed to destructive lesions of the nose and throat, respiratory tract, spleen, and kidneys. Wegener had no difficulty in identifying the underlying pathologic changes as a mixture of vasculitis and granuloma formation. He believed the condition to be related to polyarteritis nodosa, but set apart from it in some way by its distinctive clinical picture. Recent developments in immunologic research support his position. The condition resists anti-eponymic attempts to assign a formal name and continues to be known everywhere as Wegener's granulomatosis27, 41-43.
In 1949 Jacob Churg and Lotte Straus, pathologists at Mount Sinai Hospital in New York, gathered and studied 13 cases of patients who exhibited a fatal combination of severe asthma, fever, eosinophilia, necrotizing glomerulonephritis, cutaneous and subcutaneous nodular lesion, and symptoms of vascular compromise in other organ systems27,44. They suggested that the findings of granulomatous lesions within vessel walls as well as in connective tissues throughout the body set this entity apart from classical polyarteritis nodosa27, 44. This combination of signs, symptoms, and pathologic changes, which is usually designated as allergic angitis and granulomatosis, is also often called the Churg-Strauss syndrome27, 44.
In 1954, Godman and Churg reported on their evaluation of the clinical and histopathologic aspects of their cases and compared them to typical Wegener's granulomatosis15. They viewed these as a spectrum of related conditions, a concept supported by the association of these diseases and microscopic polyangiitis with ANCA15, 27, 44, 45.
Classification
One of the great challenges in medicine is the classification of vasculitis in the absence of an etiology, nonspecific signs and symptoms and few specific laboratory and imaging abnormalities. There have been a number of attempts to classify vascular disease including vascular inflammation since the mid 19th century. In 1952, Zeek put forward the classification scheme which has served as the basis for current understanding based upon vessel size and histopathology23, 24, 46. With respect to small vessel vasculitis, the designation hypersensitivity vasculitis, as used by Zeek, originally referred to disseminated necrotizing vasculitis of small arteries with frequent involvement of glomeruli, but introduced confusion in the nomenclature of small vessel inflammatory disease4.
The groundwork for many contemporary nosologica schemes was presented at the university of Texas Southwestern Medical Center by Gilliam and Smiley in 197647. They proposed a revision of this classification scheme by subdividing Zeek's existing categories. Thereafter, a number of alternative classification systems were been proposed, necessitating the formation of consensus groups to clarify the confusing terminology47-50.
Unfortunately, defining the vasculitis small-vessel is complicated by their chameleon-like nature, overlapping symptomatology and historical appellations51. Large vessel vasculitis denotes involvement of the aorta and its primary branches. Medium vessel vasculitis includes those involving vessels of both medium and small caliber including the veins, while small vessel disease affects the arterioles, venules, and capillaries. Of all the vasculitides, cutaneous leukocytoclastic vasculitis is the most difficult to classify. The terms hypersensitivity vasculitis, microscopic polyangiitis necrotizing vasculitis and cutaneous small vessel vasculitis have a11 been used in description of leukocytoclastic vasculitis related entities51.
The classification criteria currently most commonly employed are those of the American College of Rheumatology (ACR) from 1990 based upon clinical, laboratory, and histologic criteria, and those of the Chapel Hill Consensus Conferences based mainly upon histologic criteria52, 53. The 1990 ACR classification defined hypersensitivity vasculitis, whereby the sensitivity and specificity for hypersensitivity vasculitis was lowest among the vasculitides at 71% and 83.9% by traditional criteria. Palpable purpura and a maculopapular rash are undoubtedly important, though not ubiquitous features of this entity. An age range is arbitrary, histologic findings vary with disease vary with disease evolution, and an identified causative drug may or may not precipitate the disease. The histopathology of leukocytoclastic angiitis varied with time, progressing from a neutrophilic infiltrate to a monocytic infiltrate and back again54, 55. Clinically the lesions are often polymorphic, and at some stage the classic lesions become purpuric and palpable.
However, as we and others demonstrated, these classification schemes are insufficient for classifying some cases of vasculitis, particularly those involving small vessels. Further, the ACR and Chapel Hill criteria differ in numerous ways with regard to small vessel involvement in PAN, microscopic polyangiitis, Churg-Strauss syndrome, and Wegener's granulomatosis53. Another example of uncertainty in the Chapel Hill classification scheme is that of the medium vessel vasculitis, which, according to this scheme, should not involve small vessels, although the small vessel vasculitis may involve medium sized vessels26, 53, 56. The Chapel Hill consensus conference on the nomenclature of systemic vasculitis does not use the term hypersensitivity by vasculitis. Instead, the disease is classified as cutaneous leukocytoclastic angitis. Indeed, the majority of classification schemes including these entities have systemic disease with little cutaneous involvement. Some conditions such as hypersensitivity vasculitis, Henoch-Schonlein purpura, and polyarteritis nodosa are not universally recognized25, 57-59.
Development of a classification that is clinically relevant, that is useable by various specialists, and that addresses clinical features, laboratory findings, and the underlying causes of vasculitis is a goal that remains elusive. An attempt to present a working classification was has been previously presented by Jorizzo in 1993 and others60, 61. From a practical standpoint, they suggested that vasculitis be classified as either a small-vessel cutaneous vasculitis or as a large-vessel necrotizing vasculitis. The small-vessel category may be subdivided into some of the following: idiopathic hypersensitivity vasculitis, Henoch-Schônlein purpura, essential mixed cryoglobulinemia, Waldenstrôm's macroglobulinemia, urticarial vasculitis, vasculitis associated with collagen vascular diseases such as lupus erythematosus or rheumatoid arthritis, and erythema elevatum diutinum. Thus, the patient who presents with palpable purpura and a biopsy that confirms leukocytoclastic vasculitis would be diagnosed with small vessel vasculitis.
Etiologic classification
In the following, we enumerate various causes of small vessel vasculitis (Table 1). Our aim is to provide a more practical clinical approach to the diagnosis of small vessel vasculitis rather than develop yet another novel classification scheme51, 62, 63.

Histopathology
Small vessel vasculitis refers to inflammation in the walls of small vessels. Traditionally, these are cutaneous blood vessels. The classic phenotypic manifestation of small vessel vasculitis is palpable purpura; however, this is often not present. The histopathologic features of the vessel wall inflammation are important in defining the nature of the vasculitis.
Perhaps, currently, more important than the cellular characterization is the size of vessel involved. Until better histopathologic tools are developed, the anatomy of the lesion will continue to have primacy for the diagnosis of small vessel vasculitis. Involvement of arterioles, meta-arterioles, venules, and capillaries lead to many symptoms and signs, which are nonspecific with regard to pathogenesis or histopathologic subsumed in the finding "leukocytoclastic vasculitis"60, 67, 68, 74, 76, 77.
The hallmark histopathologic pattern of small vessel vasculitis is leukocytoclastic vasculitis. A lymphocytic form (in which lymphocytes predominate) has also been described. There is still not enough evidence, however, to prove that the lymphocyte pattern is truly etiologically or clinically relevant. Old lesions of small vessel vasculitis may no longer demonstrate leukocytoclastic vasculitis and may contain mainly lymphocytes around blood vessels67, 68, 78-80. This latter consideration stresses the importance of timing when taking a biopsy in a dynamic process such as the vasculitic one. In the initial phase of disease we have observed that the predominant infiltrate is with monocytes and plasmocytes, without fibrinoid necrosis or the nuclear fragments characteristic of leukocytoclastic vasculitis81.
Leukocytoclastic vasculitis is characterized by angiocentric segmental inflammation, endothelial cell swelling, fibrinoid necrosis of blood vessel walls (postcapillary venules), and a cellular infiltrate around and within dermal blood vessel walls composed largely of neutrophils showing fragmentation of nuclei (karyorrhexis or leukocytoclasia). Erythrocyte extravasation is another key feature61, 65, 77, 82. (Figures 1, 2).
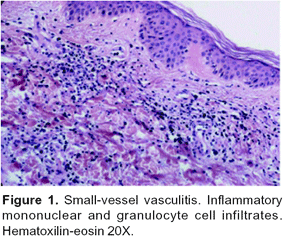
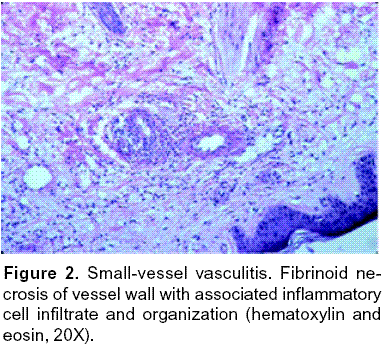
Epidemiology
The primary vasculitides are not common diseases, nor are they particularly rare. The incidence rates vary by reporting region and country. For example, rates in Europe have been detected to be between 115 and 435 per million adult populations, while secondary vasculitis occurs in about 1 per 26.5 million populations. In England, the annual incidence rate of systemic vasculitis has been reported to be 19.8 cases per million, with a prevalence of 144.5 cases per million83. In Europe, W. G. appears to be common at high latitudes, whereas PAM show the reverse pattern84.
Henoch-Schönlein purpura (HSP) is more common in children; a recent study from Spain reported an incidence of 10.5 per 100,000 children younger than 14 years of age84, 85. HSP was more common in girls, with a mean age of onset of 5.5 years, and onset was more common in autumn and winter. In 36 of cases, an upper respiratory tract infection occurred before the onset of vasculitis84, 85.
Studies of the incidence of small vessel vasculitis sui generous are complicated by the variable presentation, attribution, and lack of collaboration between physicians of different disciplines working in the field. Leukocytoclastic vasculitis comprised about 9 percent of vasculitis cases seen by rheumatologists in a study of vasculitis classification56.
Small vessel vasculitis may affect persons of any age, with a mean of about 45 years. Men and women are affected equally. LCV is the most common form of small vessel vasculitis among Caucasians in North American and Spain56, 86, 87.
One problem with using ANCA-specificity rather than the syndrome to characterize patients is that some patients with pauci-immune small vessel vasculitis are ANCA-negative, and the relationship between ANCA-specificity and clinical manifestations may be different between ethnic populations88. For example, Chen et al.89 used the Chapel Hill nomenclature system definitions and ACR classification criteria to identify 89 patients with Wegener's granulomatosis among 500 Chinese patients with ANCA _associated vasculitis. Of these 89 patients, 61% were myelopiroxidase _ANCA positive and 38% were proteinase 3 _ANCA positive. Thus in China, patients with Wegener granulomatosis more often have myeloperoxidase ANCA than proteinase _ ANCA, which is the reverse of finding in North America and Europe. Another recent epidemiological study by Gibson et al.90 in a Southern Hemisphere region, demonstrates that patients of European lineage living in New Zeland have clinical and serological profiles for Wegener's granulomatosis and microscopic polyangiitis that are very similar to Caucasian patients in Europe. That is why we set that vasculitis of small vessels can have different cutaneous and systemic phenotypic expressions and that the geographic area, race and environment contribute to the clinic heterogeneousity of the different primary vasculitis.
Etiology
Of all systemic vasculitides, vasculitis of small vessels is the form for which etiologies are best defined. Bacterial antigens may be noted in the wall of the small vessels, as may hepatitis B antigen65, 87. Still, as in all forms of vasculitis, most cases of small vessel vasculitis are idiopathic (45-54%), due to medications (10-45%), infections (10-36%), including hepatitis B (5%)65, 87, 91, 92.
Medications most commonly associated with small vessel vasculitis are antibiotics (especially â-lactams) and diuretics. Upper respiratory tract infections were the most common (20%) infectious cause of small vessel vasculitis in one series from Spain87.
Autoimmune diseases associated with secondary small vessel vasculitis include rheumatoid arthritis and systemic lupus erythematosus65, 87, 91, 92.
Phenotypic manifestations of cutaneous lesions due to small vessel vasculitis
The various entities associated with small vessel vasculitis cause similar cutaneous lesions such as palpable purpura. Central necrosis may be seen, which is indistinguishable from septic vasculitis with immune complex deposition due, for example, to gonococcal infection of that of PLEVA syndrome. Other histologic features include secondary hemorrhage, secondary microvascular thrombosis or other vasculopathy. Phenotypically, these lesions may manifest as pruritic urticarial papules, palpable purpura with central necrosis, small cutaneous nodules, rash and ulcers (Figures 3, 4). The first step is diagnosis is to recognize that small-vessel vasculitis is present, and the second more difficult step is to determine the specific type of the disease. The signs and symptoms of small-vessel vasculitis are extremely varied, and many are shared by all vessel vasculitis.
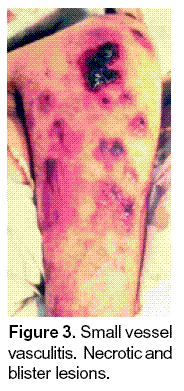
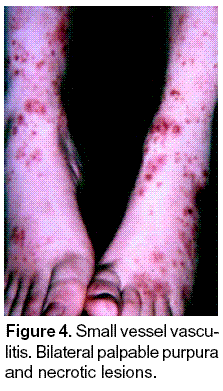
Clinical features
Cutaneous necrotizing vasculitis (small vessel vasculitis) is manifested clinically by a spectrum of cutaneous lesions, although palpable purpura is its clinical hallmark. At onset, the lesions might not be palpable, but almost all patients have purpura82. As the process continues, the lesions, which range in size from pinpoint to several centimeters, may become papulonodular, vesicular, bulbous, pustular, or ulcerated as superficial infarctions occur82. Occasionally, subcutaneous edema in the area of the vascular lesions can be observed. Lesions, usually at the same stage, occur in crops, and they appear first and predominate on the legs and ankles82. Other dependent areas under local pressure are also affected. Lesions may also occur on the other areas, but they are uncommon on the face, palms, soles, and mucous membranes. Lesions may be mildly pruritic or painful and subside within 3 or 4 weeks, leaving residual hyperpigmentation or an atrophic scar. The disease may be self-limiting, but can recur or become chronic and intermittent, with new crops of lesions appearing for months or years61, 82, 93.
Every episode of eruptive cutaneous vascular lesions may be associated with fever, malaise, arthralgia, and/or myalgia. Unusual manifestations of small vessel vasculitis may occur on dependent areas of the body or areas under local pressure or otherwise traumatized (Koebner phenomenon)52, 61, 82, 93. Clinicopathological lesions may also occur in internal organs, presumably due to circulating immune-complex-mediated vessel damage at those sites. Small-vessel vasculitis involving the nervous system may be clinically manifested by focal or diffuse, central, or peripheral neurologic involvement. Similarly, the following effects may occur: small vessel involvement of glomeruli (proteinuria or hematuria), the synovia (polyarthritis), gastrointestinal tract (abdominal pain or gastrointestinal bleeding), the pleura (pleuritis), and pericardium (symptoms of pericardial effusion). Brief mention will be made here of other types of necrotizing vasculitis54, 61, 82, 93, 94.
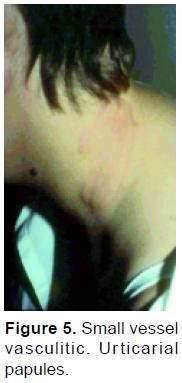
Urticarial vasculitis is characterized by wheals that persist for more than 24 hours, burn more than itch, and often leave residual purpura as they resolve82, 95 (Figure 5). Erythema elevatum diutinum is characterized by erytematosus plaques distributed symmetrically on extensor surfaces. Septic vasculitis tends to occur acrally and lesions may occasionally be wedge shaped and or papulo-pustular35, 96.
Some morphologic subtypes are considered bland. Noninflammatory cutaneous hemorrhagic microthrombosis is seen in the antiphospholipid syndrome, cold injury, cryoglobulinemia, and cryofibrinogenemia, as well as vasculopathy associated myelodysplasia, causing platelet aggregation and microthrombosis. The same findings may be seen in heparin or coumadin induced necrosis, disseminated intravascular coagulopathy, purpura fulminans, and some cases of cholesterol embolization97, 99. From an inflammatory stand point, these vasculopathic conditions are bland, due to microvascular occlusion and, in general, are not initially characterized by erythema of the cutaneous lesion. Reticular cutaneous lesions may also be associated with small vessel vasculitis. Both livedo and livedo reticularis are seen in cutaneous vasculitis due to IgA and livedoid vasculopathy. Reticular lesions may also be seen in Wegener's granulomatosis and in some cases of mixed cryoglobulinemia. These lesions are almost always erythematous at disease outset. They may be palpable and may become confluent, cause cutaneous ulcerations, and, occasionally, scarring97-99.
Cutaneous lymphocytic vasculitis
This form of vasculitis is characterized by lymphomonocytic cell infiltration as a primary response to various antigens such as medications or antigens found in connective tissue diseases such as lupus erythematosus, primary Sjögren's syndrome. These inflammatory cells are present in the involved vessel wall, while fibrinoid necrosis and leukocytoclasis is absent. Activated lymphocytes elaborate cytokines, thereby damaging the vessel wall, either by direct action of the cytokine or promotion of apoptosis. This form of vasculitis is infrequent but poorly studied57-60.
A particular form of vasculopathy associated with lymphocytic infiltrates, erythrocyte extravasation, and presence of siderophagic cells without clear evidence of vascular wall damage may be found in purpuric eruptions and lesions, Schamberg's disease, purpura of Gougerot and Blum, lupus pernio and perniosis57-59, 64, 92, 100.
Drug-induced vasculitis
Drug-Induced vasculitis should be considered in any patient with small-vessel vasculitis and will be substantiated most often in patients with vasculitis confined to the skin. Drug cause approximately 10 percent of vasculitic skin lesions. Drug-Induced vasculitis usually develops within 7 to 21 days after treatment begins 26, 101.
Acute and chronic forms of leukocytoclastic vasculitis
Erythema elevatum diutinum (EED) is characterized by papules appearing in a symmetric fashion on the extensor surface of the joints of the elbows, hands, knees, and, occasionally, the buttocks. These papular lesions often progress to larger anular lesions. Biopsy of early lesions reveals angiocentric neutrophil lymphocytoclasis. Rare deposits of fibrin may be present in the superficial and deep dermis. In addition, extravascular infiltrates with neutrophils, lymphocytes, plasmocytes, and histiocytes are present in the subdermal fatty tissues (so called cholesterolosis) or more generalized in the dermis (pandermic)57-59, 64.
Chronic stages of this lesion reveal nodular angiocentric lesions with fibrosis, eosinophilia, and capillary proliferation. It is difficult to diagnose EED in this stage, in which there may be activation of factor XIII at the level of dermal dendrocytes. Streptococcal antigens such as streptokinase and streptodornase have been implicated as causative in EED57-59, 64. Other conditions associated with EED are myelodysplastic syndromes, IgA multiple myeloma, acute myeloid leukemia, inflammatory bowel diseases, relapsing polychondritis, and rheumatoid arthritis.
Small vessel vasculitis with systemic manifestations
Henoch-Schönlein Purpura (HSP) is the most common form of systemic vasculitis in children. IgA immune complexes are present in the walls of the arterioles, venules, and capillaries. The peak incidence in children is at about five years of age. It often follows an upper respiratory tract infection. Principle manifestations of HSP are palpable purpura, arthralgias, and abdominal pain. Up to one-half of patients develop hematuria and proteinuria, while only about 10 to 20 percent of patients present with pulmonary disease and neuropathy1.
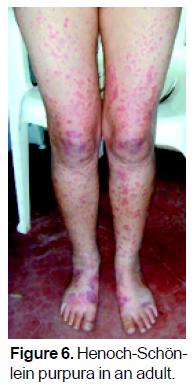
HSP can affect persons of any age. In adults, it most commonly occurs in about the third decade of life, with no gender predilection (Figure 6). Often, a precipitating antigen can be identified, such as infections or insect bites. In adults, however, the incidence of HSP and the severity of its clinical manifestations appear not to be the same as in children. Pillebout et al.102 demonstrated that clinical presentation of HSP in adults is severe and its outcome relatively poor, worse than in children. Identification of clinical and histologic prognostic factors may permit the design of appropriate therapeutic prospective studies.
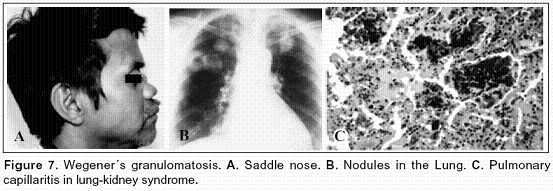
Secondary forms of small vessel vasculitis may simulate HSP. ANCA associated vasculitis may present with abdominal pain, palpable purpura, nephritis, and pneumonitis. These include microscopic polyangiitis, Wegener's granulomatosis (Figure 7), Churg-Strauss disease, and medication related ANCA-positive syndromes. The disease course may be severe, and high dose glucocorticosteroid therapy and immunosuppressive agents may be required1.
Paraneoplastic syndromes
Small vessel vasculitis may appear as a manifestation of neoplastic disease. Symptoms and signs of neoplastic disease may also simulate vasculitis. Myeloproliferative disorders and B- and T-cell lymphomas may cause lymphomonocytic or lymphocytoclastic perivasculitic infiltrates simulating vasculitis. Such cases are often difficult to correctly diagnose and are refractory to treatment1.
Diagnosis
The diagnosis and classification of patients with small vessel is based on clinical criteria, serologic as well as hispathologic; but the spectrum of small vessel vasculitis is very wide and this fact requires the doctor to pay attention to the case, it is to have in mind the fact that it may exist some limitations in the search for etiologic agents and the specificity and sensitivity of some biologic markers as P and C-ANCA, which we consider here next. In most of leucocitoclastic vasculitis, we do not have a biologic marker to classify its etiology.
The diagnosis of ANCA-associated vasculitis is made on the basis of the clinical findings, by biopsy of a relevant involved organ (typically kidney, nasal mucosa, or occasionally lung) and the presence of ANCA. Testing for ANCA using both indirect inmunofluorescence and antigen-specific enzyme linked inmunosorbent assay is recommended and provides a high sensitivity (approximately 99%) and good specificity (approximately 70%)3-14, 16, 18, 19.
How do these in vitro effects of ANCA correlate it disease activity, particularly in comparison with titles of ANCA Changes in various in vitro effects of PR3-ANCA have been suggested to follow changes in disease activity more accurately than changes in ANCA titles alone18, but this suggestion is based on observations of small numbers of patients only and so far remains to be proven. Further elucidation of the different epitopes on PR3 and MPO recognized by ANCA and their relation to disease activity may be one of the clues to this question103-107.
Evaluation of vessels in patients with Wegener's granulomatosis, microscopic polyangiitis, and Churg-Strauss syndrome revealed only a paucity of immunoglobulin deposits. This group of pauci-immune small vessel vasculitis was found to be closely associated with ANCA by serology. However, some patients have ANCA-negative. ANCA associate vasculitis103-107. Also, not all patients with ANCA-positive vasculitis have pauci-immune disease. As discussed by Hogan et al.109-110 and by Hoffman and Langford111 for some purpose it might be more appropriate to use the broad category of ANCA-associated vasculitis and to try to give a more specific syndromatic diagnosis (ie. Wegener's granulomatosis, microscopic polyangiitis, or Churg-Strauss syndrome) to classify patient management, PR3 ANCAs and MPO ANCAS are sensitive and specific markers for the idiopathic paucimmune small-vessel vasculitides and are each associated with particular clinical and histopathology112.
Is the pathogenesis of CSS different in patients who are persistently negative for ANCAs. The data from the study by Sinico et al.113 suggest that small-vessel vasculitis is generally absent in these ANCA-negative patients and that tissue infiltration by eosinophils is more prominent.
Serological and radiological test lack the sensitivity and specificity to be employed in isolation for disease assessment103 for this reason the current standard for disease assessment are clinical tools which integrate a large amount of information103.
Laboratory findings
Laboratory screening tests are always required in patients with small vessel vasculitis, (cutaneous necrotizing vasculitis) both to confirm the diagnosis and to determine the extent of systemic vasculitis, or the existence of underlying associated diseases. The necessary laboratory evaluations include histopathologic and occasionally immunofluorescent and imunophenotypical microscopic studies, blood tests, and urinalysis. The next step should be the evaluation of the systemic involvement of the patient. Complete history, physical examination, and laboratory screening are mandatory. Moreover, the identification of possible causative agents is a relevant part of patient evaluation. Three categories of etiologic factors should be considered: drugs, infectious agents, or diseases associated with increased levels of circulating immune-complexes65, 97, 99.
Examples of implicated drugs induce vasculitis are aspirin, penicillin, thiazides, and sulfonamides Examples of infective agents more often associated with necrotizing venulitis are hepatitis B, streptococcal agents, and mycobacterium tuberculosis. Diseases associated with immune-complex formation include malignancies, connective tissue diseases, inflammatory bowel disease, and chronic active hepatitis65, 97. The patient should also be evaluated for cryoglobulinemia and macroglobulinemia, collagen vascular disease, Sjogren's syndrome, lymphoma, multiple myeloma, leukemias, and solid tumors. One should realize, however, that absolute proof of etiology for a given agent is usually difficult. A large number of the above-mentioned conditions have been implicated as causes of necrotizing venulitis by temporal association, but only a few have been supported by direct evidence (ie, antigen demonstration in circulating immune complexes and in dermal (blood vessels)65, 97, 99.
Anti-neutrophil cytoplasmic antibodies (ANCA) are a heterogeneous group of autoantibodies with a broad spectrum of clinically associated diseases. ANCA testing has been established as a useful tool for the diagnosis of small vessel vasculitides, especially of `ANCA-associated vasculitides' (AAV), such as Wegener's granulomatosis, microscopic polyangiitis and Churg-Strauss syndrome, in which circulating ANCA are commonly found. Within the last 20 years these antibodies were subject of intensive studies and a growing body of evidence arose for a distinct role of ANCA in the pathogenesis of the AAV. Our current concept of whether ANCA directly or indirectly contribute to vascular damage (ANCA-cytokine-sequence-theory) was mainly developed from in vitro studies and is supported by data from clinical investigations as well as animal models. Recently a direct causal link between ANCA and the development of glomerulonephritis and vasculitis has been demonstrated. We now know that a passive transfer of ANCA is sufficient to induce disease, but it remains to be discovered how the autoantibodies to neutrophil antigens might triggered3-14, 16, 18, 19, 103-111.
The ANCA can add to it a value to the specificity to the criteria diagnoses and when combining them with the clinic and histopathologic criteria, can establish a specific diagnose in agreement to the criteria of Chapel Hill of 1994. When analyzing and observing the clinical phantom of the of small vessels vasculitis not associated to ANCA, almost they do not jeopardize the kidneys and the lungs. On the contrary, the ones associated to ANCA and the systemic vasculitis related to hepatitis C, jeopardize the kidneys and the lungs. When discriminating if the antibodies are P and C - ANCA, the prevalence of these antibodies jeopardize the kidneys at the beginning, during the course and evolution of the disease, or in the relapses of those patients with both anti-PR3 and anti-MPO positives with vasculitis, when studying them globally, the prevalence oscillates between 75% to 90% according to the study of Franssen and cols.114, 115; but if the involvement is in the respiratory tree,, anti-PR3 are detected more frequent than anti-MPO, according to the studies of Falk et al.116 and Franssen et al.114, 115. But if the skin and nervous system are involvement, anti-MPO are more frequent than anti-PR3.
One of the most interesting questions when studying this group of patients, it is to analyze the utility to follow up patients with ANCA associated vasculitis. One of the first studies was made by Tervaert et al.117 in 1989 when studying the relationship between active Wegener's granulomatosis and the titles of anti PR3. The authors observed that titles related to activity and the power to "forecast" relapses on the population studied. One year later, the same authors began to treat patients with Wegener's granulomatosis based on the elevation of anti-PR3 and thus "prevent relapse"118.
Predictors of relapse
Relapses in the vasculitis of small vessels are observed specially in AAV and complement-related vasculitis.
Differences in the pathomechanism may explain some of the variation in diseases characteristics. Are there specific features in that organ that are critical to the relapse, or are they simply organs where subclinical disease can evolve into clinically overt manifestations at an earlier stage into recognizable symptoms, signs, or serological evidence of active diseases. For example, it is easier to detect a recurrence of skin vasculitis, manifesting as a rash, than the presence of glomerulonephritis, which may only be found by careful testing of urine, assessment of renal function, and regular monitoring of blood pressure. Alternatively, some organs might be prone to flares due to a higher likelihood of infection such us nasal colonization with Saureus105. The pattern of organ involvement in a number of vasculitis is quite specific, even though in theory all vascular beds could be affected. Disease relapse occurred in 42% of patients who achieve a remission, which was within the range of previously reported relapse rated of 11% to 57%. A European study previously reported that patients with Wegener's granulomatosis were more likely to relapse than patients with microscopic polyangiitis105. Hogan et al.109, 110 demonstrate the risk for relapse was increased in the presence of anti- PR3 antibodies and lung or upper airway involvement, whereas specific disease diagnosis (W.G vs P.A.M) did not independently predict relapse. Concluded that this difference in the predictive values of specific disease diagnosis possibly reflected differences in the relative frequency of the two diseases between Europe and the southeastern United States. An association between lung involvement and relapse was informed by Kyndt et al.119 in 1999 and by Koldingsnes and Nossent120 in 2003. Previous reports did not specifically identify upper respiratory tract diseases as a risk factor for relapse; however, upper airway colonization with Saureus was associated with a higher relapse rate in patients with W.G, according to the studies of Stegeman et al.121 and Popa et al.122. According to conclusions of Hogan et al. study109, 110 female, black patients, or those with severe kidney disease, may be resistant to initial treatment more often than other patients with AAV. In a study related to outcome and prognostic factors during the course of primary Small-vessel vasculitis (P.S V.V) by Pavone et al.123 in Parma (Italy) about 75 patients, 36 with Wegener's granulomatosis (GW), 23 with Churg-Strauss syndrome (CSS), and 16 with microscopic polyangiitis (MPA), the authors conclude that Gastrointestinal (GI) involvement was associated with an increased risk of relapse, mainly in the patient with CSS, whereas renal disease and perinuclear antineutrophyl cytoplasmic antibody positivity were correlated with a lower risk of relapse. The presence of nasal Staphylococcus aureus tended to increase the risk of relapse in CSS , but to decrease it in WG123. Older age, renal and hepatic involvement, erythrocyte sedimentation rate > 100 mm /h, and serum creatinine level > 1.5 mg/ dl were all related to higher risk of death in univariate analysis; however, only cerebral, hepatic involvement and serum creatinine level > 1.5 mg / dl were independently correlated with an unfavorable prognosis for survival123. The risk of death associated with each of these indicators did not depend on the form of PSVV. The Pavone study suggests that in CSS, patient have an increased risk of relapse if there is gastrointestinal involvement, supporting the concept that some clinical feature can predict outcome123-125.
Finally, relapse is an important outcome measure in patients with AAV. Although relapses are common in these diseases, it remains unclear why these occur and whether they are influenced by exogenous or endogenous factor111 a key to minimizing the consequences of relapse is early recognition through monitoring111, 124, 125. This is particularly essential to detect glomerulonephiritis that is often asymptomatic and can be rapidly progressive. It would be important in the future to identify factors that may distinguish patients at risk of relapse or markers that reliably predicts the occurrence of relapse prior to organ injury111, 124, 125.
Treatment
Therapy of cutaneous vasculitis depends on whether or not there is clinical and laboratory evidence of internal involvement, the severity of cutaneous and systemic disease. The treatment of small vessel vasculitis, can be divided in three: Vasculitis non associated to ANCA that constitutes most of the cases in the spectrum of small vessels vasculitis; ANCA associated vasculitis and the systemic vasculitis related to the hepatitis C virus. In this paper we are going to reviews only the treatment of vasculitis associated and not associated to ANCA.
Vasculitis non- associated to ANCA
When we are facing this kind of small vessels vasculitis, we must do the identification of the antigens that induces the diseases. When the antigen is eliminated the vasculitis can be cured; for this reason, the list mentioned above must be investigated, such as medicaments, agents that simulate hemotopoiesis, vaccines, food, and leucotrieno inhibitors, modifiers of biologic response, it is not to forget that some medicaments can induce ANCA, such as herbicides, insecticides and other petroleum derives52, 56, 97.
Patients with acute cutaneous vasculitis in whom there is an identifiable cause, such as a drug, are treated symptomatically in addition to removing the presumed causative agent. Similarly, patients with Henoch-Schönlein purpura usually have self-limiting disease and are often not given specific treatment. Symptomatic measures include rest, elevation, gradient support stockings, and antihistamines. Glucocorticoides (GS) have been used in patients with renal insufficiency, specially the elderly. GS have been used in patients with severe gastrointestinal manifestations, such as abdominal pain; however, their use in the treatment of abdominal due to HSP is still controversial126. The management of HSP nephritis is also highly controversial. Patients with severe nephritis have been treated in many ways, including oral glucocorticoids or pulse therapy, alone or in combination with immunosuppressive agents such as cyclophosphamide, azathioprine, or cyclosporine126.
The challenge is to treat the patient who has chronic cutaneous vasculitis in whom there is no easily identified cause and who does not have significant systemic involvement. There is often a question regarding the need for therapy, since these patients do not have life-threatening disease. However, many of the patients have disease that alters their ability to function normally. Patients may develop small ulcerations that can become secondarily infected or may be painful. Patients may not leave their homes because of psychic distress that the presence of purpura causes. Last, patients with urticarial vasculitis complain of itching and burning of their lesions that may result in sleep disturbance.
Antihistamines have often been suggested as a first line of therapy, based on the observation that histamine may enhance the deposition of immune complexes in the vessel walls. Patients with palpable purpura rarely benefit from these agents. However, they are the corner stone of therapy for patients with urticarial vasculitis. Also, non-sedating agents may be used in the morning such as loratadine combined with a sedating antihistaminic prior to go to bed such as hydroxizine or doxepin.
For mild cutaneous involvement, 0,6 mg of colchicine two or three times daily may be helpful. Azathioprine 100-200 mg a day may be used alone or as a glucocorticoid sparing agent65 other immunosuppressive agents, including methotrexate, 10-25 mg weekly, and cyclosporine, 3-5 mg/kg per day, may be given in acute progressive disease or systemic involvement, or as steroid sparing agents65. In severe disease, either extensive cutaneous involvement or severe systemic involvement, oral prednisone 60 mg q.d. with cyclophosphamide 100 mg. q.d is effective. A gradual taper of the prednisone should be undertaken to avoid disease rebound65.
Dapsone
4,4' Diaminodiphenylsufone (DOS) is an interesting therapeutic choice for several forms of vasculitis, such as leukocytoclastic vasculitis and the urticarial vasculitis syndrome. The anti-inflammatory action of dapsone is linked to its strong quenching effect and the significant inhibition of the leukocyte respiratory burst pathway by direct suppression of the generation of toxic oxygen intermediates. It seems established that besides selectively inhibiting polymorphonuclear cytotoxicity and chemotaxis, dapsone can also inhibit mitogen-stimulated lymphocyte transformation and improve immune complex-mediated diseases127. The therapeutic potential of dapsone in vasculitis has been investigated as a monotherapy, and in combination with prednisone to allow the administration of lower doses of both drugs, thus minimizing side effects and the risk of relapse after treatment discontinuation. A daily dosage of 50 a 100 mg seems to provide a good anti-inflammatory effects. Dapsone 100 mg daily is effective in many patients with involvement restricted to the skin, and in erythema elevatum diutinum65.
Antimalarial drugs
The most widely used antimalarial drugs in dermatology include chloroquine and hydroxychloroquine. They are 4-aminoquinolones, synthetic derivatives of quinine, a naturally occurring alkaloid extracted from the bark of the South American cinchona tree. The effectiveness of antimalarials in small-vessel cutaneous vasculitis is controversial and reports are anecdotal. A larger controlled study should be performed to assess the successful indications of antimalarial therapy in cutaneous vasculitis65, 127.
Corticosteroids
Corticosteroids (CS) are the most widely used drugs for vasculitis. They can be administered alone or in combination with cytotoxic agents depending on the severity of the disease. Corticosteroids are very active as both anti-inflammatory and immunosuppressive agents. Relevant side effects can derive from long-term therapy, so combined use of CS-sparing drugs can be useful in disorders characterized by a chronic course. Corticosteroids have variable effects on cutaneous vasculitis128. They are able to improve the symptoms related to inflammation, including pain and swelling. Prednisone is usually given at the starting dose of 30 to 60 mg/ day, which is maintained until symptom relief occurs128. On the basis of clinical response and side effects the daily dosage can be reduced weekly until a lower maintenance dose is able to control the symptoms. Suspension of the treatment is possible, but relapses are frequent128.
ANCA associated vasculitis
Steroids and immunosuppressants are indicated to treat small vessel vasculitis. However, the therapeutic strategy is different from one disease to another. Treatment choice should be adapted to the predictable outcome, severity, pathogenic mechanisms and patient's general condition. In WG, Churg Strauss syndrome, and microscopic polyangiitis we have demonstrated that immunosuppressants should not be systematically prescribed. Immunosuppressants should be only prescribed in the most severe patients, when factors of poor prognosis are present. In Wegener's granulomatosis, immunosuppressants should be systematically prescribed together with steroids. The optimal treatment duration is usually of 12 months, or more for microscopic polyangeitis, and Churg-Strauss syndrome. A more prolonged treatment is mandatory in Wegener's granulomatosis, at least 18 months128-134. The new therapeutic strategies comprise also new immunosuppressants and new immunomodulating agents which could replace or be associated to the "older drugs"129, 131, 134.
References
1. Antonio Iglesias-Gamarra. Vasculitis refractarias: aspectos generales. Rev Col Reumatol 1999; 6(2): 144-160. [ Links ]
2. Iglesias-Gamarra A, Valle R, Egea E, Vásquez G, Salazar M. Análisis histórico de las vasculitis, su clasificación y propuesta para el entendimiento. Biomédica 1993;0: 38-56. [ Links ]
3. Csernok E. Anti-neutrophil cytoplasmic antibodies and pathogenesis of small vessel vasculitides. Autoimmunity Reviews 2003; 2: 158-164. [ Links ]
4. Davies DJ, Moran JE, Niall JF, Ryan GB. Segmental necrotosing glomerulonephritis with antineutrophil antibody: posible arbovirus aetiology BMJ 1982; 285: 606. [ Links ]
5. Hall JB, Wadham BM, Eood CJ, Ashton V, Adam WR. Vasculitis and glomerulonephritis: a subgroup with an antineutrophil cytoplasmic antibody. Aust N Z J Med 1984; 14: 277-278. [ Links ]
6. van der Woude FJ, Rasmussen N, Lobatto S, Wiik A, Permin H, van Es LA, et al. Autoantibodies against neutrophils and monocytes: tool for diagnosis and marker of disease activity in Wegener's granulomatosis. Lancet 1985; 1: 425-429. [ Links ]
7. Falk RJ, Jennette JC. Anti-neutrophil cytoplasmic autoantibodies with specificity for myeloperoxidase in patients with systemic vasculitis and idiopathic necrotizing and crescentic glomerulonephritis. N Engl J Med 1988; 318: 1651-1657. [ Links ]
8. Jennette JC, Hoidal JR, Falk RJ. Specificity of anti-neutrophil cytoplasmic autoantibodies for proteinasa 3. Blood 1990; 75: 2263-2264. [ Links ]
9. Jennette JC, Falk RJ. Anti-neutrophil cytoplasmic autoantibodies: discovery, specificity, disease associations and pathogenic potential. Adv Pathol Lab Med 1995; 8: 363-378. [ Links ]
10. Goldschmeding R, van der Schoot CE, ten Bokkel Huinink D, Hack CE, van den Ende ME, Kallenberg CG, et al. Wegener's granulomatosis autoantibodies identify a novel diisopropylfluorophosphate-binding protein in the lysosomes of normal human neutrophils. J Clin Invest 1989; 84: 1577-1587. [ Links ]
11. Niles JL, McCluskey RT, Ahmad MF, Arnaout MA. Wegener's granulomatosis autoantigen is a novel neutrolhil serine proteinase. Blood 1989; 74: 1888-1893. [ Links ]
12. Ludeman J, Utecht B, Gross WL. Anti-neutrophil cytoplasm antibodies in Wegener's granulomatosis recognize an elastinolytic enzyme. J Exp Med 1990; 171: 357-362. [ Links ]
13. Gross WL, Schmitt WH, Csernok E. ANCA and associated diseases: immunodiagnostic and pathogenetic aspects. Clin Exp Immunol 1993; 91: 1-12. [ Links ]
14. Kallenberg CGM, Brouwer E, Weening JJ, Cohen Tervaert JW. Antineutrophil cytolasmic antibodies: current diagnostic and pathophysiologic potential. Kidney Int 1994; 46: 1-15. [ Links ]
15. Godman GC, Churg J. Wegener's granulomatosis: pathology and review of the literature. Arch Pathol 1954; 58: 533-553. [ Links ]
16. Hoffman GS, Specks U. Antineutrophil cytoplasmic antibodies. Arthritis Rheum 1998; 41: 1521-1537. [ Links ]
17. Hagen EC, Daha MR, Hermans J, et al. Diagnostic value of standardized assays for anti-neutrophil cytoplasmic antibodies in idiopathic systemic vasculitis. EC/BCR project for ANCA assay standardization. Kidney Int 1998; 53: 743-753. [ Links ]
18. Kallenberg CG, Tervaert JW. What is new with antineutrophil cytoplasmic antibodies: diagnostic, pathogenetic and therapeutic implications. Curr Opin Nephrol Hypertens 1999; 8: 307-315. [ Links ]
19. Choi HK, Lamprecht P, Niles JL, Gross WL, Merkel PA. Subacute bacterial endocarditis with positive cytoplasmic antineutrophil cytoplasmic antibodies and antiproteinase 3 antibodies. Arthritis Rheum 2000; 43(1): 226-231. [ Links ]
20. Gamarra Iglesias A, Restrepo Suarez JF, Valle R, Osorio E, Bolaños A, Méndez O, Matteson E. Historia de las vasculitis. Rev colomb Reumato 2002; 9: 87-121. [ Links ]
21. Willan R. On cutaneous diseases. Vol. 1. London: J. Jonson, 1808: 452-471. [ Links ]
22. Bauer GG. De púrpura hemorrágica. Halae, 1824. [ Links ]
23. Zeek PM, Smith CC, Weeter JC. Studies on periarteritis nodosa III. The differentiation between the vascular lesions of periarteritis nodosa and of hypersensitivity. Am J Pathol 1984; 24: 889-917. [ Links ]
24. Zeek PM, Periarteritis nodosa: a critical review. Am J Clin Pathol 1952; 22: 777-790. [ Links ]
25. Davson J, Ball J, Platt R. The kidney in periarteritis nodosa. QJM 1948; 17: 175-202. [ Links ]
26. Jennette JC, Falk RJ, Andrassy K, Bacon PA, Churg J, Gross WL, et al. Nomenclatura of systemic vasculitides: proposal of an internacional consensus conference. Arthritis Rheum 1994; 37: 187-192. [ Links ]
27. Jennette JC, Falk RJ. Small-Vessel Vasculitis. The New England Journal of Medicine. 1997; 337: 1512-1523. [ Links ]
28. Crissey JT, Parish LC. Vasculitis: The historical development of the concept. Clin Dermatol 1999; 17: 493-497. [ Links ]
29. Schönlein JL. Allgemeine und specielle Pathologie und Therapie. 3rd ed. Vol. 2. Herisau, Switzerland: Literatur-Comptoir, 1837: 48. [ Links ]
30. Henoch E. Über den Zusammenhang von Purpura und Intestinal störungen. Berl Klin Wochenschr 1868; 5: 517-519. [ Links ]
31. Henoch EH. Lectures on diseases of children: a handbook for physicians and students. New York: W. Wood, 1882. [ Links ]
32. Osler W. The visceral lesions of purpura and allied conditions. BMJ 1914; 1: 517-525. [ Links ]
33. Heberden W. Commentarii de morborum historia et curatione. London, T. Payne, 1801. [ Links ]
34. Radcliffe-Crocker H. Erithema elevatum diutinum. Br J Dermatol 1894; 6: 1-9, 33-38. [ Links ]
35. Weidman FD, Besancom JH. Erythema elevatum diutinum. Arch Dermatol 1929; 20: 593-620. [ Links ]
36. Kussmaul A, Maier R. Ueber eine bisher nicht bescriebene eigenthümliche Arterien-erkrankung (periarteritis nodosa), die mit Morbus Brightii und rapid fortschreitender allgemeiner Muskellähmung einhergeht. Arch Klin Med 1866; 1: 484-518. [ Links ]
37. Matteson EL. A History of Idiopathic Vasculitis. Mayo Foundation for Medical Education and Research, 1999. [ Links ]
38. Matteson EL. Notes on the History of eponymic idiopathic vasculitis: the diseases of Henoch and Schönlein, Wegener, Churg and Strauss, Horton, Takayasu, Behcet, and Kawasaki. Arthritis Care Res 2000; 13: 237-245. [ Links ]
39. Matteson EL. Historical perspective of vasculitis: Polyarteritis Nodosa and Microscopic Polyangiitis. Current Rheumatolo Rep 2002; 4: 67-74. [ Links ]
40. Wohlwill F. Über die nur mikriskopisch erkennbare Form der Periarteritis nodosa. Arch Path Anat Physial Klin Med 1923; 246-377. [ Links ]
41. Groos WL, Schnabel A, Reinhold-Keller E. Wegener's. Granulomatosis: clinical Aspects In: Vasculitis edited by Gene V Ball S Louis Bridges Jr Chapter 25 Oxford. university Press, pp. 357-365. [ Links ]
42. Klinger H. Grenz formender Periarteritis nosoda. Franfurten Zeitschrift für Pathologie 1932; 42: 455-480. [ Links ]
43. Wegener F. Über eine eigenartige rhinogene Granulomatose mit besondere Beteiligung des Arteriensystems under Nieren. Beitr Pathol Anat Allg Pathol 1939; 102: 36-68. [ Links ]
44. Churg J, Strauss L. Allergic granulomatosis, allergic angiitis, and periarteritis nodosa. Amm J Pathol 1951; 27: 277-294. [ Links ]
45. Cantillo Turbay J, Iglesias, Restrepo JF. Hitos históricos de las vasculitis de pequeños vasos. Rev Colomb Reumatol 2006; 13: 142-152. [ Links ]
46. Zeek PM. Periarteritis nodosa and other forms of necrotizing angiitis. New England J Med 1953; 248: 764-772. [ Links ]
47. Gilliam, J.N. and Smiley, J.D. (1976) Cutaneous necrotizing vasculitis and related disorders. Annals of Allergy 1976; 37: 328-339. [ Links ]
48. Coperman, PWM, Ryan, T.J. The problems of classification of cutaneous angiitis with reference to histopathology and pathogenesis. British Journal of Dermatology, 1970; 82: 2-14. [ Links ]
49. Fauci, A.S., Hayness, B.F. and Katz, P. The spectrum of vasculitis; clinical, pathologic, immunologic, and therapeutic considerations. Annals of Internal Medicine 1978; 89: 660-676. [ Links ]
50. Lie, J.T. Systemic and isolated vasculitis a rational approach to classification and pathologic diagnosis. Pathology Annals 1989; 24: 25-114. [ Links ]
51. Sams HH, Sams Jr WM. Cutaneous leukocytoclastic vasculitis. In: Vasculitis Edited by Gene V Ball, & Louis Bridger Jr. Oxford University Press 2002, chapter 34, pp. 467-475. [ Links ]
52. Matteson. Historical perspective on the classification of vasculitis. Arthritis Care Res 2000; 13: 122-127. [ Links ]
53. Cantillo Turbay J, Iglesias A, Restrepo JF. Análisis crítico de las clasificaciones de las vasculitis. Rev Col de Reumatol 2006; 13: 48-64. [ Links ]
54. Sams WM Jr, Thorne EG, Small P, et al. Leukocytoclastic vasculitis. Arch Dermatol 1976; 112: 219-226. [ Links ]
55. Zax RH, Hodge SJ, Callen JP. Cutaneous leukocytoclastic vasculitis: Serial Histopahologic evaluation demonstrates the dynamic nature of the infiltrate. Arch Dermatol 1990; 126: 69-72. [ Links ]
56. Hunder GG, Arend WP, Bloch DA, Calabrese LH, Faucí AS, Fries JF, et al. American College of Rheumatology 1990 criteria for the classification of vasculitis: introduction. Arthritis Rheum 1990; 33: 1065-1067. [ Links ]
57. Neil-Crowson A, Mihm Jr MC, Magro CM. Cutaneous vasculitis: a review. J Cutan Pathol 2003; 30: 161-173. [ Links ]
58. David FF. Cutaneous vasculitis. J Am Acad Dermatol 2003; 48: 311-343. [ Links ]
59. Fiorentino DF. Cutaneous vasculitis. J Am Acad Dermatol 2003; 48: 311-340. [ Links ]
60. Jorizzo JL. Classification of vasculitis. J Invest Dermatol 1993; 100 (suppl): 106S. [ Links ]
61. Lotti T, Ghersetich I, Comacchi C, Jorozzo JL. Cutaneous small vessel vasculitis. J Am Acad Dermatol 1998; 39: 667-687. [ Links ]
62. Iglesias-Gamarra A, Valle R, Abud Mendoza C. Vasculitis de vasos de pequeño calibre. En: Tratado de Reumatología. Editores: Javier Molina, Donato Alarcón-Segovia. Nomos Ed 2007. 1121-1131. [ Links ]
63. Callen JP. Cutaneous vasculitis and Its Relation ship to systemic disease in: Inflammatory Diseases of Blood Vessel. Edited by: Gary S. Hoffman Cornelia M. Weyand Marcil Dekker INC-New York, Basel, 2002 Chapter 36, pp. 529-538. [ Links ]
64. Callen JP. Cutaneous vasculitis: Relationship to systemic disease and therapy. Curr Probl Dermatol 1993; 5: 45-80. [ Links ]
65. Sams HH, Sams WM Jr. Cutaneous leukocytoclastic vasculitis in vasculitis. Ball GV, Bridges SL Jr, eds. Oxford University Press 2002; pp. 467-475. [ Links ]
66. Mullick FC, McAllister HA, Wagher BM, Fenoglio JJ Jr. Drug related vasculitis. Human Pathol 1979; 10: 313-325. [ Links ]
67. Soter A, Mihm MC Jr, Gigli L, et al. Two distinct cellular patterns in cutaneous necrotizing angiitis. J Invest Dermatol 1976; 66: 344-350. [ Links ]
68. Soter NA. Cutaneous necrotizing venulitis. Fitzpatrik TB, Eisen AZ, Wolff K, eds. Dermatology in General Medicine Vol. 1. New York, McGraw Hill, 1993; 1501-1510. [ Links ]
69. Black AK. Urticarial vasculitis. Clin Dermatol 1999; 17: 565-569. [ Links ]
70. Wisnieski JJ. Urticarial vasculitis. Curr Opin Rheumatol 2000; 12: 24-31. [ Links ]
71. Palazzo E, Burgeois P, Meyer O, De Bandt M, Kazatchkine M, Kahn MF. Hypocomplementemic urticarial vasculitis syndrome, Jaccoud's syndrome, Valvulopathy: A new Syndrome combination. J Rheumatol 1993; 20: 1236-1240. [ Links ]
72. MacDuffie FC, Sams WM Jr, Maldonado JE, et al. Hypocomplementemia with cutaneous vasculitis and arthritis. Clin Immunol 1987; 79: 605-610. [ Links ]
73. Schwartz HR, McDeffie FC, Black LF, et al. Hypocomplementemic urticarial vasculitis: associated with chronic obstructive pulmonary disease. Mayo Clin Proc 1982; 57: 231-238. [ Links ]
74. Mehregan DR, Hall MJ, Gibson LE. Urticarial vasculitis: a histopathologic and clinical review of 72 cases. J Am Acad Dermatol 1992; 26: 441-448. [ Links ]
75. Wiesnieski JJ, Baer AN, Christensen J, et al. Hypocomplementemic urticarial vasculitis syndrome. Medicine 1995; 74: 24-41. [ Links ]
76. Woywodt A, Schneider W, Morack G, et al. Necrotizing small-vessel vasculitis confined to the uterine cervix. Semin Arthritis Rheum 2000; 29: 368-372. [ Links ]
77. Sams WW Jr, Thorne EG, Small P. Leukocytoclastic vasculitis. Arch Dermatol 1976; 112: 219-226. [ Links ]
78. Alexander EE, Moyer C, Travlos GS, Roth JB, Murphy ED. Two histopathologic types of Inflammatory vascular disease in MRL/MP autoimmune mice. Model for human vasculitis in connective tissue disease. Arthritis Rheum 1985; 28: 1146-1155. [ Links ]
79. Massa MG, Su wpd: Lymphocytic vasculitis is it a specific clinicopathologic entity? J Cutaneous Pathol 1984; 11: 132-139. [ Links ]
80. Farkas Natbony S, Phillips ME, Elias JM, Goodfrey HP, Kaplan AP. Histopathologic studies of chronic idiopathic urticaria. J Allergy Clin Immunol 1983; 71: 132-139. [ Links ]
81. Egea E, Garavito de Egea G, Severino S, Fals-Borda E, Ariza A, Munar W, Iglesias Gamarra A. Vasculitis linfomonocítica Informe de nueve casos. Acta Médica Colombiana 1987; 12: 330-338. [ Links ]
82 Ghersetich I, Comachic, Jorizzo JL, Katsambas A, Lotti TM. Proposal for a working classification of cutaneous vasculitis. Clinics in Dermatology 1999; 17: 499-503. [ Links ]
83. Watts RA, Carruthers DM, Scott DGI. Epidemiology of systemic vasculitis: changing incidence of definition. Sem Arthritis Rheum 1995; 25: 28-34. [ Links ]
84. Watts RA, Scott D GI. Epidemiology of the vasculitis. Current Opinion Rheumatology 2003; 15: 11-16. [ Links ]
85. Calvino MC, Llorca J, Garcia-Porrua C, Fernández-Iglesias JL, Rodriguez-Ledo P, González-Gay MA. Henoch-Schönlein purpura in children from northwester Spain: a 20 year epidemiologic and clinical study. Medicine (Baltimore) 2001; 80: 279-290. [ Links ]
86. Ekenstam E, Callen JP. Cutaneous leukocytoclastic vasculitis: clinical and laboratory features of 82 patients seen in private practice. Arch Dermatology 1984; 120: 484-489. [ Links ]
87. Martínez-Taboada VM, Blanco R, García-Guente M, Rodriguez-Valverde V. Clinical features and outcome of 95 patients with hypersensitivity vasculitis. Am J Medicine 1997; 102: 186-191. [ Links ]
88. Jennette JC, Falk RJ. Nosology of primary vasculitis. Curr Opin Rheumatol 2007; 19: 10-16. [ Links ]
89. Chen M, Yu F, Zhang Y, Zou WZ, Zhao MH, Wang HY . Characteristics of Chinese patients with Wegener's granulomatosis with antimyeloperoxidase autoantibodies. Kidney Int 2005; 68: 2225-2229. [ Links ]
90. Gibson A, Stamp LK, Chapman PT, O'Donnell JL. The epidemiology of Wegener's granulomatosis and microscopic polyangiitis in a Southern Hemisphere region. Rheumatology (Oxford) 2006; 45: 624-628. [ Links ]
91. Callen JP, Chandra JJ, Voorhees JJ. Cutaneous angiitis (vasculitis). Int Dermatol 1978; 17: 105-108. [ Links ]
92. Hautmann G, Campanile G, Lotti TM. The many faces of cutaneous vasculitis. Clin Dermatol 1999; 51: 31-37. [ Links ]
93. Lotti T, Comacchi C, Ghersetich I. Cutaneous necrotizing vasculitis. Int J Dermatol 1996; 35: 457-474. [ Links ]
94 Jorizzo JL, Solomon AR, Zanolli MD, et al. Neutrophilic vascular. Arch Dermatol 1976; 112: 219-216. [ Links ]
95. Gammon WR. Urticarial vasculitis. Dermatol Clin 1985; 3: 97-105. [ Links ]
96. Katz SI, Gallin JU, Hertz KC, Fauci AS, Lawley TJ. Erythema elevatum diutinum: Skin and systemic manifestations, immunologic studies and successful treatment with dapsone. Medicine 1977; 56: 443-455. [ Links ]
97. Piette WW. Primary Systemic vasculitis. In: Cutaneous manifestations of Rheumatic Diseases. Editors Richard D. Sontheimer, Thomas T. Provost. Lippincott Williams & Wilkins. Chapter 8. Second Edition, pp. 159-196. [ Links ]
98. Callen JP. Cutaneous vasculitis: relationship to systemic disease and therapy. Curr Probl Dermatol 1993; 5: 45-80. [ Links ]
99. Piette WW. The differential diagnosis of purpura from a morphologic perspective. Adv. Dermatol 1994; 9: 3-24. [ Links ]
100. Le Boit PE. The true and the near-true. Am J Dermatopathol 2002; 24: 267-269. [ Links ]
101. Ekenstam E, Callen JP. Cutaneous leukocytoclastic vasculitis: clinical and laboratory features of 82 patients seen in private practice. Arch Dermatol 1984; 120: 484-489. [ Links ]
102. Pillebout E, Thervert E, Hill G, Alberti C, Vanhille P, Nochy D. Henoch-Schönlein purpura in adults: Outcome and prognostic factors. J. Am Soc Nephrol 2002; 13: 1271-1278. [ Links ]
103. Mukhtyar CB, Flossmann O, Luqmani RA. Clinical and biological assesment in systemic necrotizing vasculitides: Clin Exp Rheumatol 2006; 24 (2 suppl 41): S92-S99. [ Links ]
104. Kamesh L, Harper L, Savage C.O.S. ANCA-positive vasculitis J Am Soc Nephrol 2002; 13: 1593-1960. [ Links ]
105. Luqmani RA, Flossmann O. Outcome in small-vessel systemic vasculitis J Rheumatol 2006; 33: 1224-1227. [ Links ]
106. Savige J, Pollock W, Trevisin M. What do antineurophil cytoplasmic antibodies (ANCA) tell us? Best Pract Res Clin Rheumatol 2005; 19(2): 263-276. [ Links ]
107. Seo P, Stone J. The Antineutrophil cytoplasmic Antibody. Associated Vasculitides. Am J Med 2004; 117: 39-50. [ Links ]
108. Kallenberg C, Rarok A, Stegeman C, Limburg P. New insights into the pathogenesis of antineutrophil cytoplasmic autoantibody-associated vasculitis. Autoimmun Rev 2002; 1: 61-66. [ Links ]
109. Hogan SL, Nachman PH, Wilkman AS, Jennette JC, Falk RJ. Prognostic markers in patients with antineutrophil cytoplasmic autoantibody associated microscopic polyangeitis and glomerulophritis J. Am Sc Nephrol 1996; 7: 23-32. [ Links ]
110. Hogan SL, Falk RJ, Chin H, Cai J, Jennette CE, Jennette JC, et al. Predictors of relapse and treatment resistance in antineutrophil cytoplasmic antibody associated small-vessel vasculitis. Ann Intern Med 2005; 143: 621-631. [ Links ]
111. Hoffmann GS, Langford C. Are there different forms of life in the antineutrophil cytoplasmic antibody universe? Ann Intern Med 2005; 143(9): 683-685. [ Links ]
112. Kallenberg CGM. Churg-strauss syndrome: Just one disease entity? Arthritis Rheum 2005; 52: 2589-2593. [ Links ]
113. Sinico RA, Di Tama L, Maggiore O, Bottero P, Radice A, Tosoni C, et al. Prevalence and clinical significance of antineutrophil cytoplasmic antibodies in Churg-Strauss syndrome. Arthritis Rheum 2005; 52: 2926-2935. [ Links ]
114. Franssen CF, Gans R, Kallenberg C, Hageluken C, Hoorntije S. Disease spectrum of patients with antineutrophil cytoplasmic autoantibodies of defined specificity: distinct differences between patients with anti-proteinase 3 and anti-myeloperoxidase autoantibodies. J Intern Med 1998; 244: 209-216. [ Links ]
115. Franssen CF, Stegeman CA, Kallenberg CG, Gans RO, De Jong PE, Hoorntje SJ, et al. Antiproteinase 3- and antimyeloperoxidase-associated vasculitis. Kidney Int 2000; 57: 2195-2206. [ Links ]
116. Falk RJ, Hogan S, Carey TS, Jennette JC. Clinical course of antineutrophil cytoplasmic autoantibody-associated glomerulonephiritis and systemic vasculitis. The Glomerular Disease Collaborative Network. Ann Intern Med 1990; 113: 656-663. [ Links ]
117. Tervaert JW, van der Woude FJ, Fauci AS, Ambrus JL, Velosa J, Keane WF et al. Association between active Wegener's granulomatosis and anticytoplasmic antibodies. Arch Intern Med 1989; 149: 2461-2465. [ Links ]
118. Tervaert JW, Huitema MG, Hené RJ, Sluiter WJ, The TH, van der Hem GK, et al. Prevention of relapses in Wegener's granulomatosis by treatment based on antineutrophil cytoplasmic antibody titre. Lancet 1990; 336: 709-711. [ Links ]
119. Kyndt X, Reumaux D, Bridoux F, Tribout B, Bataille P, Hachulla E, et al. Serial measurements of antineutrophil cytoplasmic autoantibodies in patients with systemic vasculitis. Am J Med 1999; 106: 527-533. [ Links ]
120. Koldingsnes W, Nossent JC. Baseline features and initial treatment as predictors of remission and relapse in Wegener's granulomatosis. J Rheumatol 2003; 30(1): 80-88. [ Links ]
121. Stegeman CA, Tervaert JW, Sluiter WJ, Manson WL, de Jong PE, Kallenberg CG. Association of chronic nasal carriage of Staphylococcus aureus and higher relapse rates in Wegener granulomatosis. Ann Intern Med 1994; 120: 12-17. [ Links ]
122. Popa ER, Stegeman CA, Boss NA, Kallenberg CG, Tervaert JW. Staphylococcal superantigens and T cell expansions in Wegener's granulomatosis. Clin Exp Immunol 2003; 132: 496-504. [ Links ]
123. Pavone L, Grasselli C, Chierici E, Maggiore U, Garini G, Ronda N, et al. Secondary and Primary Vasculitides (Se. Pri.Va) Study Group. Outcome and prognostic factors during the course of primary small-vessel vasculitides. J Rheumatol 2006; 33; 1299-1306. [ Links ]
124. Lurati-Ruiz F, Spertini F. Predictive value of antineutrophil cytoplasmic antibodies in small-vessel vasculitis. J Rheumatol 2005; 32(11): 2167-2172. [ Links ]
125. Stegeman CA. Predictive value of antineutrophil cytoplasmic antibodies in small vessel vasculitis: Is the Glass half full or half empty? J Rheumatol 2005; 32: 2075-2077. [ Links ]
126. Gonzalez-Gay MA, Garcia-Porria C. Henoch-Schönlein purpura. In: Vasculitis. Edited by Gene V. Ball, Louis Bridges J.R Chapter 25. Oxford. University Press, 476-494. [ Links ]
127. Atzori L, Ferreli C, Biggio P. Less common treatment in cutaneous vasculitis. Clin Dermatol 1999; 17: 641-647. [ Links ]
128. Vena G, Cassano N. Immunosuppressive therapy in cutaneous vasculitis. Clin Dermatol 1999; 17: 633-640. [ Links ]
129. Molloy E, Langford C. Advances in the Treatment of small vessel vasculitis. Rheum Dis Clin North Am 2006; 32: 157-172. [ Links ]
130. Goek O, Stone J. Randomized controlled trial in vasculitis associates with anti-neutrophil cytoplasmic antibodies. Curr Opin Rheumatol 2005; 17: 257-264. [ Links ]
131. Levy J. New aspect in the management of ANCA-positive vasculitis. Nephrol Dial Transplant 2001; 16: 1314-1317. [ Links ]
132. Uthman I. Pharmacological therapy of vasculitis: an update. Curr Opin Pharmacol 2004; 4: 177-182. [ Links ]
133. Langford C, Ballow J. New insights into the immunopathogenesis and treatment of small vessel vasculitis of the kidney. Curr Opin Nephrol Hypertens. 2003; 12(3): 267-272. [ Links ]
134. Guillevin L, Pagnoux C. When Should Immunosuppressants Be Prescribed to Treat Systemic Vasculitides? Intern Med 2003; 42: 313-317.
[ Links ]
