










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Reumatología
Print version ISSN 0121-8123
Rev.Colomb.Reumatol. vol.15 no.2 Bogotá Apr./June 2008
Raquitismo hipofosfatémico ligado al X (XLH): Presentación de una familia, asociado a una osteoartritis prematura y simulando además una espondiloartropatía seronegativa
X-linked hypophosphatemic rickets: Presentation of a family, associated with premature osteoarthritis, and simulating a seronegative spondyloarthropathy.
Gerardo Quintana López1, José Félix Restrepo2, álvaro Sánchez3, Enrique Calvo4, Andrés Fernández1, Antonio Iglesias Gamarra5
1Médico Internista y Reumatólogo. Departamento de Medicina, Facultad de Medicina. Universidad Nacional de Colombia, Sede Bogotá.
2Médico Internista y Reumatólogo. Profesor Titular. Facultad de Medicina. Universidad Nacional de Colombia. Jefe Departamento Médico, Facultad de Medicina. Universidad Nacional de Colombia, Sede Bogotá.
3Médico Reumatólogo. Profesor asociado. Facultad de Medicina. Unidad de Reumatología. Universidad Nacional de Colombia, Sede Bogotá.
4Médico Radiólogo. Profesor asistente. Facultad de Medicina. Universidad Nacional de Colombia, Sede Bogotá.
5Médico Internista y Reumatólogo. Profesor titular. Facultad de Medicina. Unidad de Reumatología. Universidad Nacional de Colombia, Sede Bogotá.
Recibido para publicación: febrero 15/2008. Aceptado en forma revisada: mayo 22/2008
Resumen
En este artículo presentamos un enfoque práctico para el diagnóstico diferencial de desórdenes hipofosfatémicos heredados junto a la osteomalacia inducida por tumor (una forma adquirida), profundizando sobre el raquitismo hipofosfatémico ligado al cromosoma X y hacemos la descripción de una familia con este diagnóstico.
Palabras clave: raquitismo hipofosfatémico ligado a X, espondiloartropatía.
Summary
In this article we present a practical focus for the differential diagnosis of hypophosphatemic disorders inherited join to the osteomalacia induced by tumor (an acquired form); deepening about the X-linked hypophosphatemic rickets and we present description of a family with this diagnosis.
Key words: X-linked hypophosphatemic rickets, spondyloarthropathy.
Introducción
El crecimiento y desarrollo del esqueleto requiere adecuada suplencia de calcio y fosfato, y la deficiencia de estos minerales resulta en un deterioro de la mineralización ósea. Los defectos genéticos que llevan a la disminución de la reabsorción tubular de fosfato y la hipofosfatemia crónica son las causas más comunes de raquitismo heredado.
Dentro de los síndromes hipofosfatémicos encontramos cuatro variantes a saber: raquitismo hipofosfatémico ligado al cromosoma X (XLH del inglés X-linked hypophosphatemic rickets), raquitismo hipofosfatémico autosómico dominante (ADHR del inglés autosomal dominant hypophosphatemic rickets), la osteomalacia inducida por tumor (TIO del inglés tumor-induced osteomalacia) y el raquitismo hipofosfatémico hereditario con hipercalciuria (HHRH del inglés hereditary hypophosphatemic rickets with hypercalciuria)1,2.
En sus características químicas, los niveles de calcio sérico son normales, excepto en el HHRH que puede estar normal o elevado, el calcitriol característicamente se encuentra normal y algunas veces bajo, y elevado en el HHRH; la PTH está normal o ligeramente elevada en el XLH y TIO, normal en ADHR y suprimida en el HHRH. En todos el máximo tubular para el fosfato en relación a la tasa de filtración glomerular (TmP / GFR) se encuentra disminuido, la calciuria es normal, menos en HHRH que se encuentra elevada1-5.
En las características clínicas también hay diferencias, el XLH es la forma más frecuente, los defectos dentarios sólo se presentan en el XLH (defectos de la dentina y abscesos) y en ADHR (abscesos dentarios); la debilidad muscular es mínima en XLH, prominente en TIO y presente en ADHR y HHRH. En cuanto a la herencia el XLH es dominante ligada a X, el ADHR es autosómica dominante, la TIO está asociada a tumor y el HHRH es autonómica recesiva; la penetrancia se describe como completa con expresión variable en el XLH, incompleta en la ADHR y variable en el HHRH. El defecto cromosómico está en el Xp22.1 en XLH, en 12p13 en el ADHR y desconocido en el HHRH. El gen defectuoso en el XLH es el PHEX (Phosphate regulating gene with homologies to endopeptidase on the X chromosome), en el caso del ADHR y TIO es el FGF- 23 (fibroblast growth factor) y desconocido en el HHRH1-11. Ver Tabla 1.
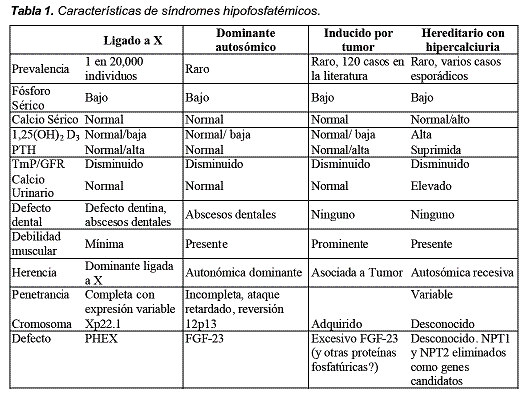
Raquitismo hipofosfatémico ligado a X
El XLH fue descrito por primera vez en el año de 1937 por Albright y cols.12. Es el desorden hipofosfatémico heredado más común, representando el 80% de los casos, es un desorden hereditario dominante ligado al X con poco o ningún efecto de dosis genética con una prevalencia de 1: 20.000, antiguamente llamado raquitismo resistente a la vitamina D o hipofosfatemia ligada al cromosoma X1-4.
La hipofosfatemia ocurre como consecuencia de una reabsorción tubular renal de fósforo inorgánico disminuida; el máximo tubular para el fosfato en relación con la tasa de filtración glomerular (TmP / GFR) se encuentra reducido1,2. El XLH es causado por mutación en el gen PHEX, el cual codifica para una metaloproteasa, la M1313. Se ha descrito un factor humoral como la causa de pérdida renal de fosfato a partir de observaciones hechas en ratones14 y también por la evidencia mostrada luego de transplantar pacientes con insuficiencia renal crónica con XLH y recurriendo inmediatamente la fosfaturia 15. El origen de tal factor circulante es desconocido pero de los estudios en modelos animales se ha sugerido que el osteoblasto puede producir el factor16, 17. Pero la expresión no solo se realiza en el osteoblasto, sino también en los osteocitos, odontoblastos, músculos, pulmones y ovarios, por ello se explica las diferentes alteraciones que se observa en los pacientes con XLH. Además se ha sugerido la presencia de un daño intrínseco en el osteoblasto que contribuiría a la enfermedad ósea en el XLH1. Evidencias adicionales sugieren que la mutación del gen PHEX produce un depósito y expresión anormal de la matriz ósea18. Se han descrito otras mutuaciones puntuales a nivel del PHEX en algunas cepas de ratones, que se caracterizan por la pérdida de fosfatos, hipofosfatemia y raquitismo, así mismo también se han identificado más de 180 mutaciones del PHEX en numerosas familias XLH y en casos individuales. (www.phexdb.mcgill.ca). El defecto en el osteoblasto y la evidencia de un factor humoral han llevado a la hipótesis de que el defecto óseo intrínseco lleva a la liberación de factores humorales que afectan la mineralización ósea, la síntesis de calcitriol y la absorción tubular de fosfato1.
Se caracteriza por presentar una baja estatura, raquitismo con la resultante deformidad en las extremidades inferiores, dolor óseo, fracturas o pseudofracturas y entesopatía (calcificaciones de tendones, ligamentos y cápsulas articulares)1-4,19. Frecuentemente, estos pacientes desarrollan osteoartritis, así como también ha sido informada la compresión de médula espinal en adultos no tratados4. No hay alteración de la función intelectual ni de la expectativa de vida. Adicionalmente, se encuentra niveles aumentados de fosfatasa alcalina1-4.
Los pacientes pediátricos presentan cambios radiológicos de raquitismo activo, con excavaciones, engrosamiento y/o desgaste en los extremos metafisiarios de los huesos largos. Existe también pérdida de la zona provisional de calcificación y engrosamientos de la placa epifisiaria. Las metáfisis presentan una estructura trabeculada2.
En cuanto al tratamiento, se hace con suplencia de fosfato junto con calcitriol, para evitar el hiperparatiroidismo. Así, se revierte el raquitismo, descienden los niveles de fosfatasa alcalina y la tasa de crecimiento reaparece2. Se ha descrito también, el uso de diuréticos como hidroclorotiazida y amiloride20.
Se presenta la primera familia observada durante 36 años, donde analizamos la presencia de raquitismo hipofosfatémico y el progreso de la osteoartritis que simulan una espondiloartropatía seronegativa.
Materiales y métodos
Se estudió la familia B durante 36 años. C.J. de 68 años, el caso índice, sus tres hijos (dos gemelos P.B y O.B de 42 años, y uno de 38 años BM) y un hermano (L) fallecido. A todos se les practicaron Rx simple de cráneo, huesos largos, pelvis, columna, manos y rodillas, estudios de calcio, fósforo en sangre y orina de 24 horas, fosfatasa alcalina total, albúmina, globulina y solo a dos (C.J.B. y P.B) estudios de PTH y calcitriol.
Resultados
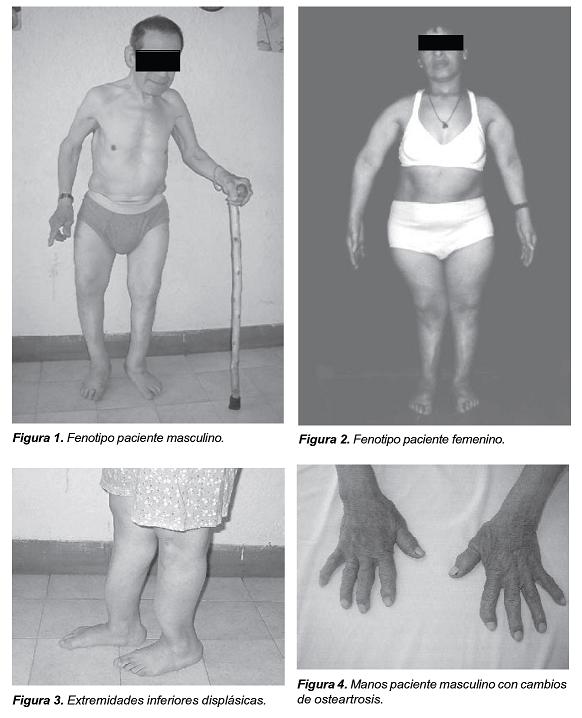
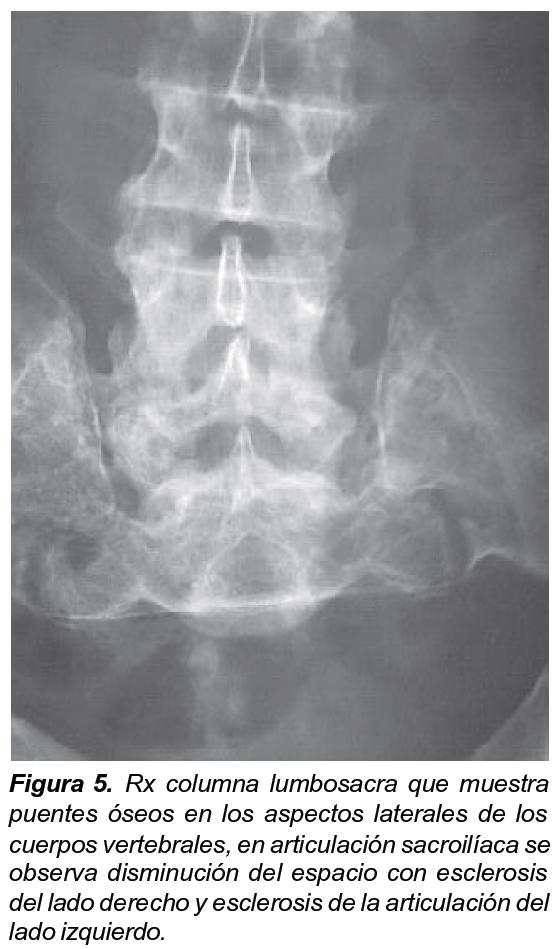
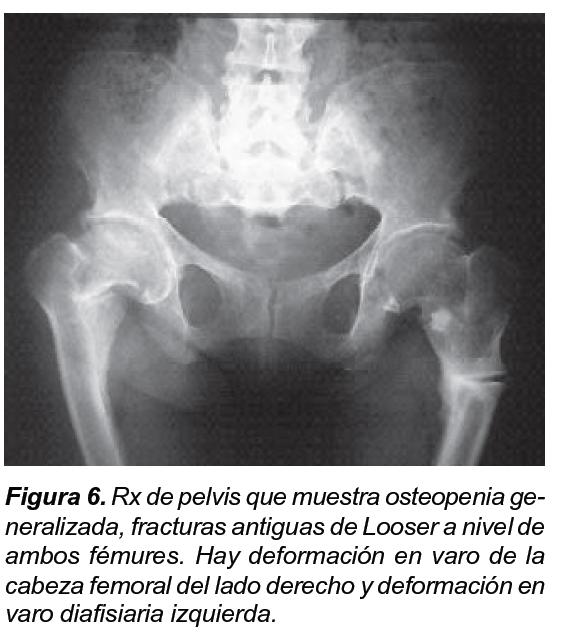
Los niveles de fósforo oscilaron entre 0,6, 1,8 y 2 (n= 3-4,5 mg/dl) y la fosfaturia en orina de 24 horas entre 400 y 1500 mg en 24 horas (n= 400 – 1300 mg/dl), los niveles de fosfatasa alcalina oscilaron entre 212 y 4000 (VN 30-50 en 1966). Los niveles de calcitriol y PTH en los pacientes estudiados fueron normales. Se observaron las siguientes características: alteración de la dentina, dientes anchos y deformados, tórax protruido en su cara anterior, codos y puños ensanchados, caderas en aducción y rotación externa, y deformación de miembros inferiores (Figuras 1-4). Los RX son compatibles con osteomalacia en el adulto, con osteopenia, formación de sindesmofitos, parecidos a los que se observan en la espondiloartropatía (Figuras 5 y 6). A partir de los 30 años los pacientes desarrollan una osteoartritis prematura, que es generalizada y progresiva que los lleva a la incapacidad, especialmente para la marcha y de la movilidad de la columna.
Discusión
Los síndromes hipofosfatémicos heredados son un grupo de enfermedades muy poco frecuentes y dentro de estas el XLH es el más prevalente (80%). Consideramos que el caso que se presenta constituye un diagnóstico claro de esta entidad, correspondiendo a una forma ligada a cromosoma X y heredado en la descendencia; también queremos llamar la atención en cuanto a la presencia de formación de sindesmofitos con osteopenia, hallazgos que simulan una espondiloartropatía y en la literatura informada encontramos muy pocos casos similares: la primera referencia es del año 1978, hecha por Nelson y cols.21 quienes describen cuatro casos de osteomalacia con compromiso axial atípico, dentro de los cuales dos simulan una espondilitis anquilosante; posteriormente Reginato y Cols.22 en una serie de casos, 26 en total, describen la osteomalacia y sus manifestaciones osteomusculares, dentro de las cuales solo uno simula una espondilitis anquilosante. Finalmente, Akkus y cols. describen un caso de osteomalacia en una mujer de 39 años con una sintomatología sugestiva de espondilitis anquilosante, pero al realizar el estudio de laboratorio y de imágenes se pudo confirmar el diagnóstico23.
Así, ante la presencia de la enfermedad se debe iniciar un estricto control y tratamiento, y vigilancia de la descendencia para determinar la presencia de hipofosfatemia en la edad pediátrica e iniciar su tratamiento, y en caso de mutaciones nuevas se encontrará detención del crecimiento y se debe iniciar el estudio para este tipo de patología, con calcio y fósforo séricos y en orina de 24 horas, niveles de paratohormona, fosfatasa alcalina y estudio radiológico.
Conclusión
Por efecto del gen PHEX se produce una inadecuada degradación / inactivación de un factor fosfatúrico como la fosfatina. Esta hormona produce una represión de la expresión genética del cotransportador NPT2, con la consiguiente pérdida renal de fosfato y la hipofosfatemia, lo que conlleva al raquitismo en los niños y la osteomalacia en el adulto.
Se describe, entonces, un XLH con osteoartritis temprana, que puede simular una espondiloartropatía seronegativa. Es la primera familia descrita en Colombia y en Latinoamérica con 36 años de seguimiento.
Referencias
1. Jan De Beur SM, Levine M. Molecular Pathogenesis of Hypophosphatemic Rickets. J Clin Endocrinol Metab 2002; 87: 2467-2473. [ Links ]
2. Carpenter T. New perspectives on the biology and treatment of X-linked hypophosphatemic rickets. Pediatr Clin of North Am 1997; 44: 443-466. [ Links ]
3. Pronicka E, Popowska E, Rowinska E, Piekutowska D, Oglecka M, Krajewska- M, et al. Biochemical and DNA markers of X-linked hypophosphatemic rickets: A study of sporadic cases. J Inher Metab Dis 1992; 15: 335-338. [ Links ]
4. Reid IR, Hardy DC, Murphy WA, Teitelbaum SL, Bergfeld MA, Whyte MP. X-linked hypophosphatemia: A clinical, biochemical and histopathologic assessment of morbidity in adults. Medicine 1989; 68: 336-352. [ Links ]
5. Drezner MK. Tumor-associated rickets and osteomalacia. In Favus MJ (ed): Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism, ed 2. New York, Raven Press, 1993: 282-288. [ Links ]
6. Econs MJ, McEnery PT, Lennon F, Speer MC. Autosomal dominant hypophosphatemic rickets is linked to chromosome 12p13. J. Clin Invest 1997; 100: 2653-2657. [ Links ]
7. The ADHR Consortium. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF 23. Nat Genet 2000; 26: 345-348. [ Links ]
8. Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, et al. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci USA. 2001; 98: 6500-6505. [ Links ]
9. Moncrieff MW. Early biochemical findings in familial hypophosphatemic hyperphosphaturic rickets and response for treatment. Arch Dis Child 1982; 57: 70-72. [ Links ]
10. Teider M, Modai D, Samuel R, Arie R, Halabe A, Bab I, Gabizon D, Liberman UA. Hereditary hypophosphatemic rickets with hypercalciuria. N Engl J Med 1985; 312: 611-617. [ Links ]
11. McWhorter AG, Seale NA: Prevalence of dental abscess in a population of children with vitamin D-resistant rickets. Pediatr Dent 1991; 13: 91. [ Links ]
12. Albright F, Butler AM, Bloomberg E. Rickets resistant to vitamin D therapy. American J Dis Child 1937; 54: 529. [ Links ]
13. The HYP Consortium. A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. Nat Genet 1995; 11: 130-136. [ Links ]
14. Lajeunesse D, Meyer RA, Hamel L: Direct demonstration of a humorally mediated inhibition of renal phosphate transport in the Hyp mouse. Kidney Int 1996; 50: 1531-1538. [ Links ]
15. Morgan JM, Hawley WL, Chenoweth AI, Retan WJ, Diethelm AG. Renal transplantation in hypophosphatemia with vitamin D-resistant rickets. Arch Intern Med 1974; 134: 549-552. [ Links ]
16. Ecarot-Charrier B, Glorieux FH, Travers R, Desbarats M, Bouchard F, Hinek A. Defective bone formation by transplanted Hyp mouse bone cells into normal mice. Endocrinology 1998; 123: 768-773. [ Links ]
17. Rifas L, Dawson LL, Halstead LH, Roberts M, Avioli LV l. Phosphate transport in osteoblasts from normal and X-linked hypophosphatemic mice. Calcif Tissue Int 1994; 54: 505-510. [ Links ]
18. Miao D, Bai X, Dibyendu P, McKee MD, Karapalis AC, Goltzman D. Osteomalacia in Hyp mice is associated with abnormal Phex expression and with altered bone matrix protein. Endocrinology 2001; 142: 926-937. [ Links ]
19. Polisson RP, Martinez S, Khoury M, Harrell RM, Lyles KW, Friedman N, et al. Calcification of entheses associated with X-linked hypophosphatemic osteomalacia. N Engl J Med 1985; 313: 1-6. [ Links ]
20. Alon U, Chan JCM. Effects of hydrochlorothiazide and amiloride in renal hypophosphatemic rickets. Pediatrics 1985; 75: 754-763. [ Links ]
21. Nelson AM, Riggs BL, Jowsey JO. Atypical axial osteomalacia: report of four cases with two having features of ankylosing spondylitis. Arthritis Rheum 1978; 21: 715-719. [ Links ]
22. Reginato AJ, Falasca GF, Pappu R, McKnight B, Agha A. Musculoskeletal manifestations of osteomalacia: report of 26 cases and literature review. Semin Arthritis Rheum 1999; 28: 287-304. [ Links ]
23. Akkus S, Tamer MN, Yorgancigil H. A case of osteomalacia mimicking ankylosing spondylitis. Rheumatol Int 2001; 20: 239-242. [ Links ]

