










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Reumatología
Print version ISSN 0121-8123
Rev.Colomb.Reumatol. vol.16 no.1 Bogotá Jan./March 2009
ARTÍCULO DE REFLEXIÓN
Osteocondensant diseases. A new clinical-radiological vision supported in genetics and osteoimmunology
Jimi Mejía-Vallejo1, Enrique Calvo2, José Félix Restrepo3,
Antonio Iglesias-Gamarra3
1 Internista Reumatólogo, Universidad Nacional de Colombia.
2 Radiólogo, Profesor Asociado de Radiología.
3 Internista y Reumatólogo, Profesor Titular de Reumatología
Recibido: Noviembre 25/2008 Aceptado: Febrero 2/2009
Resumen
Las alteraciones en el remodelado óseo llevan al incremento o disminución de la masa ósea, generando daño de la microarquitectura ósea, lo cual incrementa el riesgo de fractura. Las patologías con incremento de la densidad conducen a diversos procesos osteocondensantes genéticamente dirigidos. La osteocondensación es explicada actualmente por alteración en la función del osteoclasto asociada a una deficiente función de la resorción ósea, alteración en la función del osteoblasto que genera un incremento anormal en la formación ósea, o un imbalance homeostático entre los dos procesos; la expresión clínica y radiológica de estas entidades puede darse en etapas tempranas del desarrollo o en la vida adulta dependiendo del componente autosómico recesivo o dominante respectivamente. En esta revisión, se discute la clasificación basada en el desorden funcional de las células óseas y las principales características clínicas y radiológicas que permiten un abordaje diagnóstico sencillo y aplicable en la práctica clínica.
Palabras clave: osteoesclerosis, hiperostosis, osteoclasto, osteoblasto.
Summary
The alterations in osseous remodeling lead to the increase or decrease of the osseous mass, generating damage to the osseous micro-architecture, which increases the risk of fracture. The pathologies with increase in osseous density lead to different genetically directed osteocondensing processes. The osteocondensing is currently explained by alteration in the function of the osteoclast, associated with a deficient function of the osseous resorption, an alteration in the osteoblast function, which generates an abnormal increase in the osseous formation, or a homeostatic imbalance between the two processes; the clinical and radiological expression of these diseases can take place in early stages of the development, or in the adult life, depending on the recessive or dominant autosomic component, respectively. In this review, the classification based on the functional disorder of the bone cell is discussed, as well as the main clinical and radiological characteristics than permit a simple and applicable diagnostic approach in the clinical practice.
Key words: osteosclerosis, hyperostosis, osteoclast, osteoblast.
Introducción
Las patologías osteocondensantes son desórdenes poco frecuentes que afectan el esqueleto axial y apendicular, caracterizados por incremento de la masa ósea e imágenes de hiperdensidad en las radiografías simples. Su etiología parece obedecer a polimorfismos genéticos que condicionan alteraciones en vías de señalización en el osteoclasto, el osteoblasto o en ambas células con desequilibrio dinámico temporo-espacial en la unidad de remodelado óseo; no obstante el mecanismo molecular y genético de algunas entidades osteocondensantes permanece en estudio. El incremento de la masa ósea involucra al hueso cortical (hiperostosis) y al hueso trabecular (osteoesclerosis) en diferentes proporciones, dependiendo de la patología y su curso evolutivo.
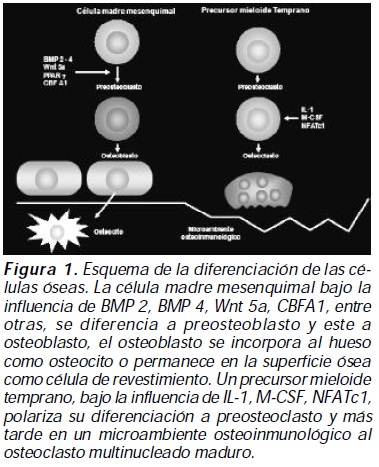
La diferenciación a osteoblastos y a osteoclastos a partir de células madre mesenquimales y de células precursoras mieloides respectivamente, es controlada por diferentes factores que son determinantes en las vías de señalización intracelular; así en presencia de BMP 2 y 4, Wnt5a, CBFA1, PPAR gama (ver tabla de abreviaturas), la polarización de la célula madre mesenquimal es hacia el osteoblasto; en tanto que en presencia de IL-1, M-CSF, y factor de transtripción NFATc1, la polarización de la célula precursora mieloide es hacia el osteoclasto1 (figura 1).
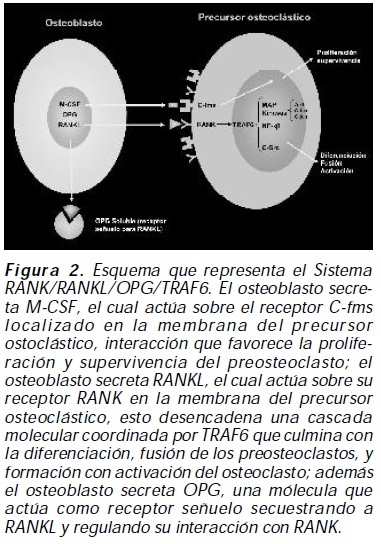
La resorción del hueso mineralizado y la degradación enzimática de la matriz ósea orgánica necesitan del funcionamiento coordinado de vías de señalización en el osteoclasto que permitan la acidificación en la laguna de resorción y la producción de colagenasas como la catepsina K; de forma similar vías de señalización en el osteoblasto regulan la formación ósea; simultáneamente el equilibrio entre la resorción y la formación ósea es controlado por sistemas de señalización dinámicos como el sistema RANK/ RANKL/OPG/TRAF6, el TGF-Beta 1, entre los más estudiados2 (figura 2).
Clasificación
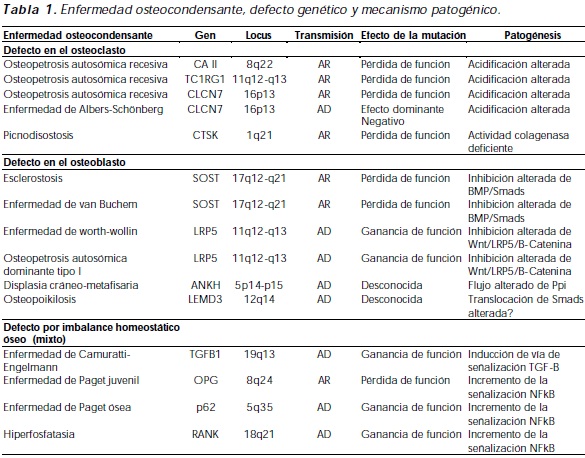
Muchas clasificaciones han sido realizadas en los pasados 50 años fundamentadas en hallazgos radiológicos, histológicos y clínicos, clasificaciones que influenciadas por la gran proliferación de subvariedades de entidades osteocondensantes no totalmente definidas o estudiadas, y múltiples epónimos crearon caos y confusión3; el continuo avance en el conocimiento en genética, biología molecular, señalización intracelular e intercelular y el desarrollo de la inmunoosteología han permitido tener conceptos más claros que facilitan enormemente su clasificación haciendo de esta útil y aplicable en la orientación clínico-radiológica. Una nueva propuesta para entender y agrupar las enfermedades osteocondensantes es la siguiente (tabla 1):
1. Enfermedades osteocondensantes por defecto en el osteoclasto
El osteoclasto es una célula gigante multinucleada responsable de la resorción del tejido óseo; el fracaso en el reclutamiento del osteoclasto hacia la unidad de remodelado óseo y su disfunción resortiva puede ser secundaria a un defecto extrínseco al osteoclasto con expresión génica defectuosa de proteínas en células presentes en el microambiente óseo que orientan su reclutamiento o por mutaciones y polimorfismos genéticos intrínsecos del osteoclasto que se traducen en la elaboración de proteínas no funcionales.
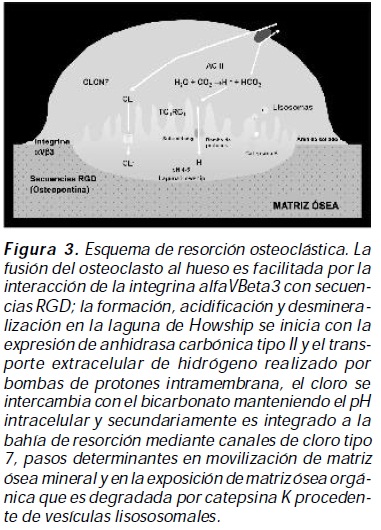
La resorción ósea es un proceso coordinado que inicialmente implica unión del osteoclasto activado al tejido óseo seguido por la resorción; así la integrina alfaVbeta 3 expresada en el osteoclasto durante su maduración, al interactuar con secuencias RGD (secuencia Arg-Gly-Asp de aminoácidos) presentes en proteínas de matriz ósea como la osteopontina, sialoproteína, vitronectina, entre otras4, establece el área de sellado óseo; la membrana plasmática toma una forma dentada, iniciándose la resorción ósea y la formación de la laguna de resorción o laguna de Howship5.
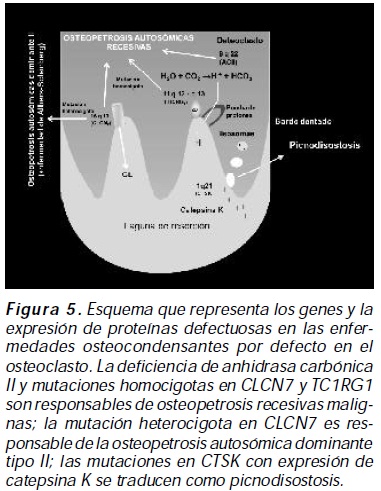
El microambiente en la laguna de resorción es acidificado hasta obtener un pH de 4 a 5 por transporte extracelular de hidrógeno realizado por bombas de protones intramembrana, el cloro se intercambia con el bicarbonato manteniendo el pH intracelular y secundariamente es integrado a la laguna de resorción mediante canales de cloro tipo 7; dos procesos determinantes en movilización de matriz ósea mineral (principalmente hidroxiapatita) y en la exposición de matriz ósea orgánica que es degradada por catepsina K procedente de vesículas lisososomales6 (figura 3). La alteración en las vías de señalización intracelular con acidificación y actividad colagenasa deficiente o defectuosa se manifiestan en diferentes formas de osteopetrosis o picnodisostosis, respectivamente.
Osteopetrosis
Las osteopetrosis incluyen un grupo heterogéneo de condiciones caracterizadas por incremento de la densidad ósea debido a resorción alteada; las diferentes subformas son clasificadas con base en defectos moleculares genéticos, edad de inicio y características clínicas en dos formas principales: la forma maligna infantil u osteopetrosis autosómica recesiva y la forma benigna adulta u osteopetrosis autosómica dominante tipo II (enfermedad de Albers-Schönberg)7 .
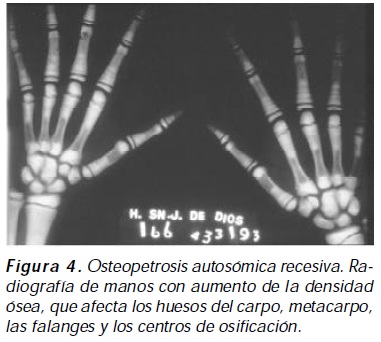
Osteopetrosis autosómica recesiva: obedece a mutaciones inactivantes en tres genes con loci identificados (tabla 1); sin embargo, mutaciones genéticas en actual estudio, como mutaciones recesivas de RANKL, podrían explicar otras formas de osteopetrosis8,9; se manifiesta durante la infancia, los pacientes suelen presentar hematopoyesis ineficaz, con esplenomegalia, hemólisis y sangrados a causa del excesivo tejido óseo en los espacios medulares, erupción retardada de la dentición, fracturas, y compresión de pares craneales con sintomatología dependiente de la estructura comprometida; además de hipertensión endocraneal, el compromiso neurológico está presente en algunos pacientes independiente de la compresión nerviosa; los huesos son densos a la exploración radiológica, comprometiendo mayormente estructuras dependientes de osificación endocondral (Figura 4). Sin tratamiento, el cual es trasplante de médula ósea, los pacientes usualmente fallecen en la primera década de la vida10 .
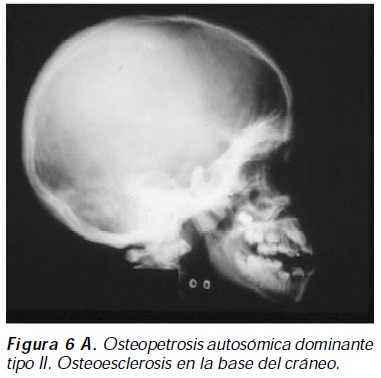
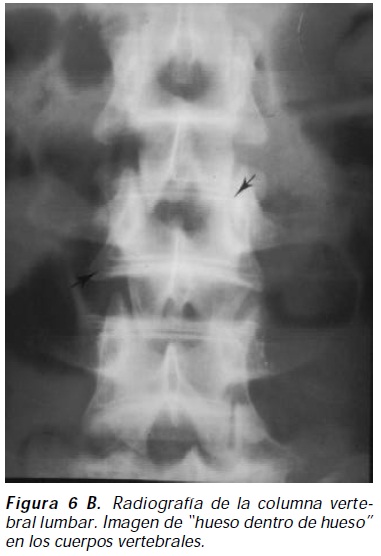
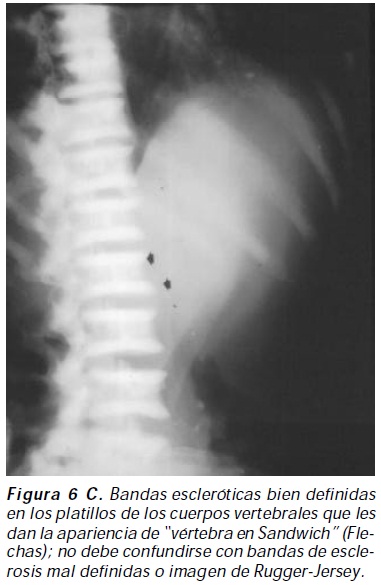
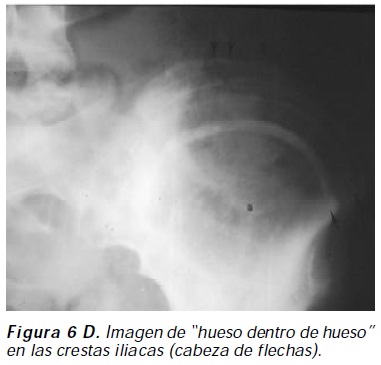
Osteopetrosis autosómica dominante tipo II (enfermedad de Albers-Schönberg): denominada también forma adulta, benigna, resulta de la pérdida de actividad del canal de cloro 7 por mutación inactivante heterocigota en el gen CICN7 con penetrancia incompleta11 (figura 5). Los pacientes suelen ser asintomáticos, las manifestaciones clínicas son escasas y ocurren usualmente en adolescentes; las principales complicaciones afectan el esqueleto, las fracturas ocurren en un 80% de los casos con una media de tres fracturas por paciente; las fracturas pueden presentarse en cualquier hueso largo y en los arcos posteriores de la vértebras, siendo el fémur el hueso más frecuentemente afectado; la osteoartrosis, osteoesclerosis de la base del cráneo, osteomielitis, hematopoyesis ineficaz y absesos dentarios son otras complicaciones informadas12 ; las características radiológicas incluyen bandas escleróticas en los platillos de los cuerpos vertebrales que dan la apariencia de "vértebra en Sandwich" casi patognomónico de osteopetrosis, bandas de esclerosis mal definidas en los platillos vertebrales o imagen de Rugger- Jersey, apariencia de "hueso en el interior de hueso" visibles en huesos iliacos, base del cráneo y cuerpos vertebrales13 (Figuras 6 A, B, C, D); las características radiográficas pueden ser identificadas de forma temprana en la infancia, con empeoramiento radiológico en el tiempo14. La consolidación defectuosa posquirúrica y la infección son comunes y su pronóstico es bueno.
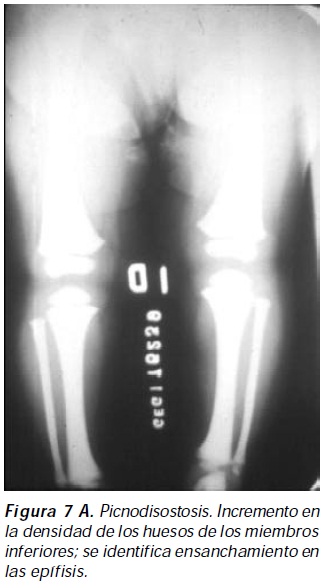
Picnodisostosis
Es la enfermedad que sufrió el pintor expresionista francés Moulin Rouge Henri de Toulouse Lautrec (1864-1901).
Posterior a la resorción de la matriz mineral ósea, el segundo paso implican degradación de la matriz ósea orgánica; la catepsina K es una enzima lisosomal ácida secretada en la laguna de resorción para fragmentar al colágeno tipo 1, a la osteopontina y la osteonectina a un pH bajo; las mutaciones en el gen CTSK que codifica para la catepsina K han sido identificadas como la causa de la picnodisostosis15.
La picnodisostosis es un trastorno autosómico recesivo generalmente diagnosticado en la infancia, con características fenotípicas propias caracterizadas por talla corta (1,35 a 1,50 metros), facies típica con cabeza grande, región frontal y occipital protuberante, cráneo alargado, nariz delgada, ángulo de la mandíbula obtuso con apariencia de micrognatia, ectopia dentaria, platibasia y paladar ojival16; tórax estrecho con pectus excavatum, cifoescoliosis, hiperlordosis lumbar, genu valgo (secundario a fracturas recurrentes en miembros inferiores); las manos y los pies son pequeños, cuadrados y con dedos cortos; las fracturas comienzan a presentarse en la segunda década de la vida17 (Figuras 7A A 7B). Los hallazgos radiológicos precisan el diagnóstico; existe osteoesclerosis generalizada, los huesos largos presentan hiperostosis con estrechamiento parcial del canal medular, acroosteólisis, las vertebras son densas con sus apófisis transversas respetadas, la calota y la base del cráneo son escleróticos con órbitas radiodensas, suele existir pérdida de la neumatización de los huesos paranasales18. Su tratamiento, paradójicamente, consiste en el uso de bifosfonatos además de hormona de crecimiento.
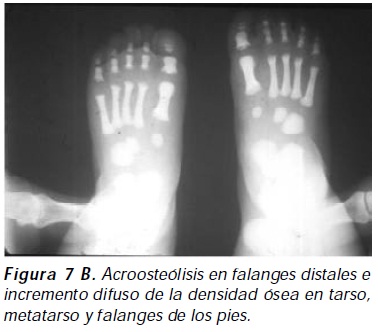
2. Enfermedades osteocondensantes por defecto en el osteoblasto
El osteoblasto es la célula formadora de hueso, ella produce matriz orgánica compuesta principalmente por colágeno tipo 1, la cual es posteriormente mineralizada; su función es condicionada por vías de señalización meticulosamente reguladas y coordinadas; parte de su desequilibrio se manifiesta con incremento de la masa ósea que se exterioriza con diversos fenotipos clínicos de enfermedades osteocondensantes; han sido identificadas cuatro anomalías genéticas que dirigen su desarrollo.
Vía de señalización BMP/Smads: las proteínas morfogénicas del hueso (BMP) son un grupo de moléculas de señalización que pertenecen a la superfamilia de péptidos TGF beta; son potentes factores capaces de estimular la diferenciación osteoblástica y la formación ósea; sus acciones en el osteoblasto son mediadas por receptores específicos de superficie celular, las proteínas Smad y las MAPKinasa, y moduladas por proteínas extra e intracelulares mediante mecanismos locales de feedback (retroalimentación negativa). Los antagonistas de BMPs bloquean las señales de transducción a múltiples niveles que incluyen antagonismo extracelular, bloqueo de sus receptores, proteínas inhibitorias intracelulares y ubiquitinización con degradación en el inmunoproteosoma de BMPs19. La unión de BMP a sus receptores fosforila a Smad 1, Smad 5 y Smad 8 (denominadas R-Smads); estas forman un complejo con Smad 4 (denominado Co-Smad) que se transloca al núcleo e interactúa con factores transcripcionales, interacción facilitada por proteínas de la membrana nuclear interna como MAN120, 21 (figura 8), para inducir la expresión génica e iniciar la formación de proteínas de la matriz ósea22 ; Smads 6 y 7 (denominadas ISmads) son mediadores de regulación negativa en esta vía de formación ósea (figura 5).
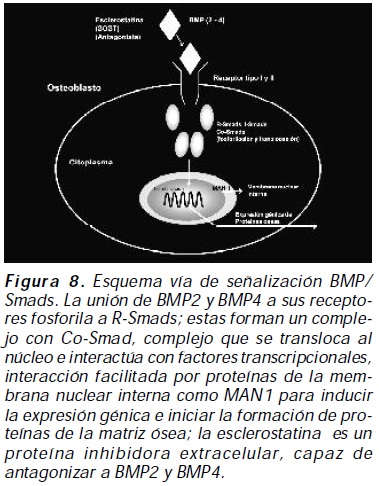
La esclerostatina o SOST es un miembro de la familia de proteínas inhibidoras extracelulares de BMP nominada Dan, expresada casi exclusivamente en el osteocito; SOST es capaz de antagonizar a BMP2 y BMP4 quizá con mediación de I-Smads23.
Vía de señalización Wnt/LRP5/ beta-catenina: esta vía juega un papel relevante en la diferenciación del osteoblasto y el mantenimiento de la masa ósea. En esta vía intervienen proteínas reguladoras, entre ellas LRP5, la cual es un correceptor putativo de membrana que tiene función dual; así facilita la formación ósea mediante la unión a proteínas denominadas Wnt y Frizzled, formando un complejo trimolecular que activa a Dsh, generando dispersión del complejo de destrucción de la beta-catenina (APC + Axin + GSK3 + CK1 alfa); así, beta-catenina se transloca al núcleo, activa factores de transcripción e inicia la expresión génica de proteínas de matriz ósea (figura 9). LRP5 con función dual inhibe coordinadamente la formación ósea mediante la unión a un antagonista extracelular denominado Dkk1 (tabla de abreviaturas), y a Kremen, una proteína de inhibición transmembranaria; la internalización citosólica de este complejo inhibe a Dsh y favorece la integración del complejo de destrucción de la betacatenina, la cual es ubiquitinizada, degradada y ulteriormente desintegrada en el inmunoproteosoma. Proteínas como SFRPS se comportan como antagonistas señuelos extracelulares, y WIF 1 ligan a Wnts e impiden su unión a LRP524, 25 .
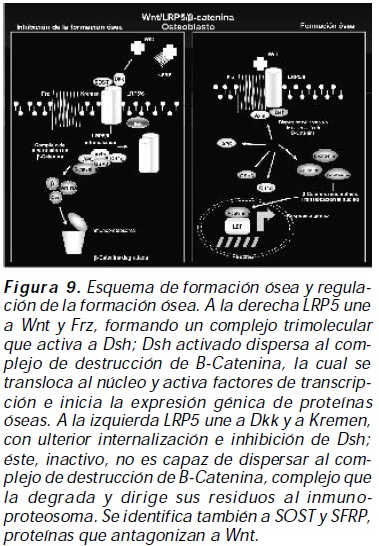
La mineralización de la matriz ósea inorgánica es regulada por el pirofosfato, un inhibidor de la formación de cristales de hidroxiapatita, el cual se desplaza hacia el espacio extracelular cooperado por múltiples proteínas transmembrana denominadas ANK que regulan los niveles de pirofosfato en el osteoblasto y son codificadas por el gen ANKH 26, 27 (figura 10).
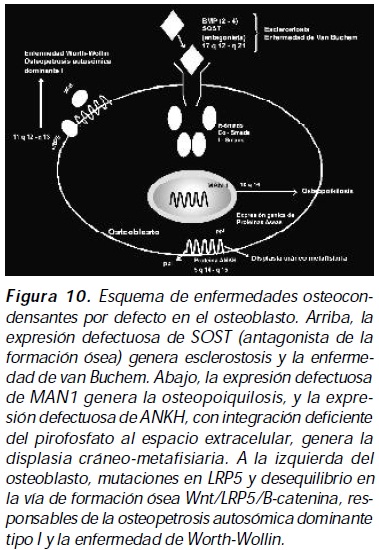
Enfermedad de van Buchem y esclerostosis (enfermedad de Truswell-Hansen)
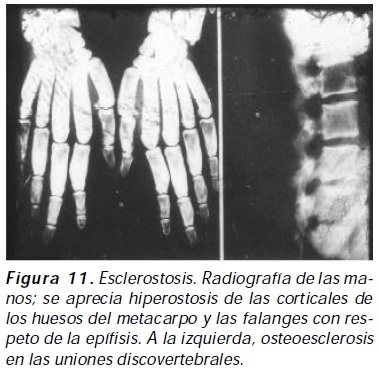
Son trastornos autosómicos recesivos generados por variaciones alélicas en el gen que codifica para SOST con repercusión en la vía de señalización BMP/Smads; cuatro mutaciones inactivantes han sido identificadas para la enfermedad de Truswell-Hansen: tres mutaciones introducen un codón de parada prematura, más una cuarta que implica la sustitución de una base y la expresión de SOST defectuosa con inapropiado empalme genético; la enfermedad de van Buchem es secundaria a una deleción 52 kb con la expresión de SOST defectuosa28. Estas dos displasias congénitas se caracterizan por engrosamiento óseo progresivo, con hiperostosis y osteoesclerosis cráneo-tubular generalizada con respeto de las epífisis, elongamiento asimétrico progresivo de la mandíbula, compromiso de neuroforámenes en la base del cráneo, dolor en huesos largos a la presión y ausencia de fracturas; la estenosis del canal medular es la principal característica radiológica, existiendo engrosamiento cortical dependiente del endostio (aunque la hiperotosis dependiente del periostio también está presente); la obliteración del espacio diploico con esclerosis simétrica del cráneo y la neumatización de senos paranasales normal son otras características radiológicas; el esqueleto axial es menos afectado, con osteoesclerosis homogénea del cuerpo vertebral y de los procesos espinosos29. La esclerostosis es diferenciada del van Buchem por la presencia de sindactilia entre el segundo y tercer dedo de las manos, displasia en uñas e incremento anormal del peso y la talla que dan la apariencia de gigantismo30 (Figura 11).
Enfermedad de Worth-Wollin
Es un transtorno autosómico dominante generado por mutación heterocigota de LRP5 V171 con alteración en la inhibición normal de la vía de señalización Wnt/LRP5/beta-catenina31; LRP5 mutado no interactúa con el antagonista Dkk1, fracasando la inhibición de beta-catenina y predominando la formación ósea32,33. Este fenotipo de síndrome de alta masa ósea tiene presentación clínica benigna; el dismorfismo facial iniciado en la adolescencia no es constante, al igual que el compromiso neurológico; en todos los pacientes se observa un torus palatinus prominente; no está asociado a fracturas; radiológicamente se caracteriza por hiperostosis cráneo-tubular; el compromiso axial es menos marcado, con incremento homogéneo de la densidad vertebral.
Displasia cráneo-metafisiaria
Es una forma rara de displasia ósea. Posiblemente la primera descripción de esta patología se hizo en un fémur y una tibia desenterrados por Smith y Jones de un cementerio nubiano en 190234. Obedece a un trastorno autosómico dominante generado por mutaciones inactivantes en ANKH con abolición o disminución marcada del flujo hacia el espacio extracelular de pirofosfato e incremento en la mineralización ósea35 (Figura 10). Se caracteriza por hiperostosis difusa simétrica progresiva del cráneo y ensanchamiento metafisiario con articulaciones normales; puede ser detectada en las primeras semanas de vida por anomalías respiratorias secundarias a senos nasales estrechos; el continuo engrosamiento de los huesos craneofaciales causa estrechamiento de los forámenes craneales, incluido el foramen magnum, llevando a condiciones como ceguera, sordera y parálisis facial; otros hallazgos clínicos incluyen dolicocefalia, hipertelorismo, nariz aplanada, mandíbula prominente, dentición de mala calidad, mala oclusión dental, extremidades desproporcionadamente largas con limitación en la extensión de codos y genu valgo. Los hallazgos radiológicos pueden ser identificables desde etapas muy tempranas; estos son caracterizados por hiperostosis difusa de la base del cráneo, bóveda craneal; el fenotipo en los huesos largos consiste en metáfisis ensanchadas con líneas de Erlenmeyer, adelgazamiento cortical, aspectos más evidentes en la parte distal del fémur y la tibia36,37 (Figura 12 ). Su valoración y tratamiento son interdisciplinarios.

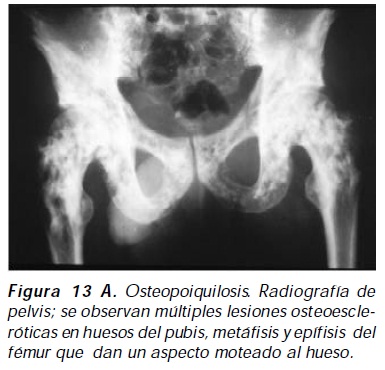
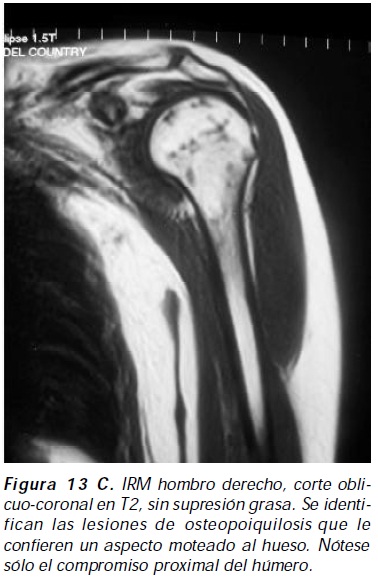
Osteopoiquilosis
La osteopoiquilosis es un desorden esclerosante autosómico dominante, secundario a mutaciones inactivantes heterocigotas en el dominio LEMD3 del gen que codifica para MAN1, proteína de la membrana nuclear interna con función reguladora postranscripcional de R-Smad, I-Smad, Smad 2 y 3; MAN1 mutada altera la inhibición normal realizada por SOST y mediada por Smads en la vía de señalización BMP/Smads38 . Se desarrolla en la adolescencia, es asintomática y se caracteriza por el hallazgo casual en radiografías simples con múltiples lesiones osteoescleróticas que dan un aspecto moteado al hueso, lesiones identificables también en la IRM (Figuras 13 A, B, C); los sitios más frecuentemente afectado son las epífisis y las metáfisis de los huesos tubulares largos, las muñecas, los tobillos, la pelvis y las escapulas, el cráneo está indemne y excepcionalmente se comprometen los cuerpos vertebrales, las costillas y el maxilar inferior; puede acompañarse de lesiones en la piel tipo dermatofibrosis lenticularis disseminata, denominándose síndrome de Buschke-Ollendorff a esta coexistencia39; otras lesiones como la escleroderma lineal han sido también informadas40; la osteopoiquilosis puede ser una expresión fenotípica más del síndrome de microdeleción del cromosoma 12q14, junto a retardo mental leve y corta estatura41. Su diagnóstico diferencial debe incluir el mieloma múltiple y las metástasis osteoblásticas por cáncer de próstata o glándula mamaria42.

3. Enfermedades osteocondensantes por imbalance homeostático óseo (mixto)
El estrecho equilibrio entre la resorción y la formación ósea es controlado por hormonas sistémicas (paratohormona y vitamina D3) que interactúan con factores locales aislados u organizados en complejos sistemas dinámicos; entre ellos el más estudiado es el sistema RANK/ RANKL/OPG/TRAF6; citoquinas proinflamatorias como la Interleuquina 1, el TNF alfa, interleuquina 6, interleuquina 17 (potenciadores de la pérdida ósea por inducción de expresión de RANKL), interleuquina 5, interleuquina 10, interleuquina 12, interleuquina 18 e interferones alfa, beta y gama (inhibidores de osteoclastogénesis por inhibición de RANKL) , y otros factores como los miembros de la superfamilia del TGF-beta (con efecto óseo dual), GM-CSF, los cuales mantienen la compleja homeostasis osteoinmunólogica43. El aumento de la masa ósea en este grupo de patologías es secundario a incremento en la función del osteoblasto, con disminución en la función del osteoclasto o incremento en la función de estas dos células; el prototipo de esta alteración es la displasia diafisiaria progresiva o enfermedad de Camurati- Engelmann.
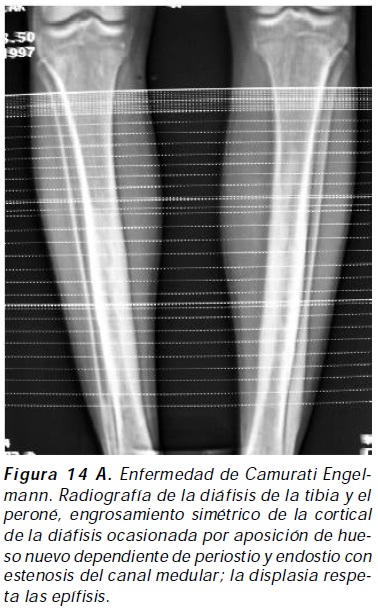
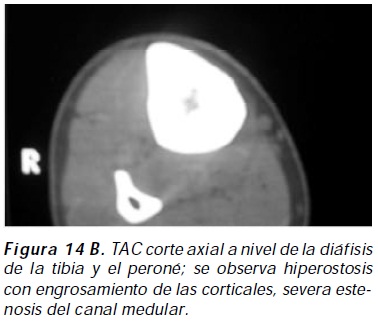
Enfermedad de Camurati-Engelmann
Esta condición osteocondensante es conocida como displasia diafisiaria progresiva. Es un desorden autosómico dominante con abundante TGF beta 1 almacenado en la matriz ósea, el cual tiene modulación negativa en la expresión de RANKL y modulación positiva en la expresión de OPG en el osteoblasto44; se caracteriza por hiperostosis gradual craneotubular que afecta el periostio y el endostio, suele acompañarse de fatigabilidad fácil, debilidad con atrofia muscular y severo dolor muscular y esquelético en miembros inferiores que obligan a adoptar al paciente una posición de gateo, mimetizando distrofia muscular en adolescentes con probable remisión en la edad adulta45,46; radiológicamente se caracteriza por engrosamiento simétrico y progresivo de la cortical de la diáfisis ocasionada por aposición de hueso nuevo dependiente de periostio y endostio, engrosamiento que puede afectar además la metáfisis, el cráneo y la pelvis; las epífisis son respetadas47,48. Se han encontrado niveles incrementados de fosfatasa alcalina y osteocalcina49. Los corticoides, la fisioterapia y los bifosfonatos forman la base de su tratamiento, mientras que los analgésicos no esteroideos son ineficaces en control del dolor50. (Figuras 14 A,B).
Conclusión
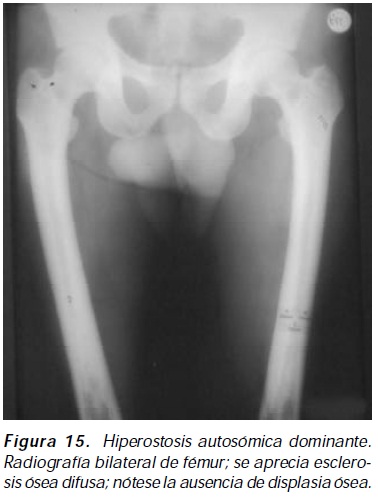
Aún existen enfermedades osteocondensantes en las cuales se ignora el mecanismo molecular que explique su expresión fenotípica, haciendo diferir su inclusión en un grupo específico de clasificación; no obstante, el continuo interés y descubrimiento en el campo de la osteoinmunología y la genética permite caracterizar y clasificar cada vez mejor estas entidades, posibilitando al reumatólogo, al endocrinólogo, al ortopedista y al radiólogo tener una visión más aproximada y fundamentada de estas complejas entidades; así, nuevas propuestas patológicas como osteopetrosis autosómica dominante tipo I51 y la hiperostosis autosómica dominante52 (Figura 15) parecen tener un horizonte cada vez más claro.
En la aproximación diagnóstica el médico osteólogo se enfrenta a una gran diversidad de enfermedades óseas (genéticas, adquiridas, metabólicas, oncológicas, metastásicas y hallazgos óseos casuales), lo que se convierte en un reto diagnóstico no siempre fácil de resolver; sin embargo, el conocer el desarrollo perinatal del individuo, la edad de la expresión clínico-radiológica, identificar compromiso de sistema nervioso central, de pares craneales, la presencia o ausencia de displasia ósea, identificar disformisfo craneofacial, historia de fracturas previas (las enfermedades osteocondensantes por defecto en el osteoblasto generalmente no están asociadas a fracturas), antecedentes familiares y el excluir causas secundarias de osteocondensación con una tamización de laboratorio básica (cuadro hemático, BUN, creatinina, calcio y albúmina séricos, fosfato sérico, AST, ALT, antígeno prostático específico, ácido úrico, electroforesis de proteínas, paratohormona, fosfatasa alcalina, VSG, PCR), radiología simple de huesos y densitometría ósea; ayudas diagnósticas cuya solicitud está siempre condicionada a una alta sospecha clínica, se convierten en importantes detalles que conducen a la exactitud diagnóstica en este complejo, desafiante e interesante grupo de enfermedades óseas.
Tabla de abreviaturas
Referencias
1. Walsh M, Kim N, Kadono Y, Rho J, Lee S, Lorenzo J, et al. Osteoimmunology: Interplay Between the Immune System and Bone Metabolism. Annu Rev Immunol 2006; 24: 33-63. [ Links ]
2. Quinn J, Itoh K, Udagawa N, Hausler K, Yasuda H, Shima N, et al. Transforming growth factor B affects osteoclast differentiation via direct and indirect actions. J Bone Miner 2001; 16: 1784-1794. [ Links ]
3. Greenspan A. Sclerosing bone dysplasias -a targetsite approach. Skelatal Radiol 1991; 20: 561-583. [ Links ]
4. Duong L, lakkakorpi P, Nakamura I, Rodan G. Integrins and signaling in osteoclast function. Matrix Biol 2000; 19: 97-105. [ Links ]
5. Takagi J, Petre B, Walz T, Springer T. Global conformational rearrangements in integrin extracellular domans in outside-in and inside-out signaling. Cell 2002; 110: 599-611. [ Links ]
6. Teitelbaum S. Osteoclast: What Do They Do and How Do They Do it?. Am J Pathol 2007; 170(2): 427-435. [ Links ]
7. Frattini A, Orchanrd P, Sobacchi C, Giliani S, Abinum M, Mattsson J, et al. Defects in TCIRG1 subunit of the vacuolar proton pump are responsible for a subset of human autosomal recessive osteopetrosis. Nat Genet 2000; 25: 343-346. [ Links ]
8. Balemans W, Van Wesenbeeck L, Van Hul W. A Clinical and Molecular Overview of the Human Osteopetrosis. Calcif Tissue Int 2005; 77: 263-274. [ Links ]
9. Tolar J, Teitelbaum S, Orchard P. Osteopetrosis. N Engl J Med 2004; 351: 2839-2849. [ Links ]
10. Frattini A, Pangrazio A, Susani L. Chloride channel CICN7 mutations are responsable for severe recessive, dominant, and intermediate osteopetrosis. The J Bone and Miner Res 2003; 18: 1740-1747. [ Links ]
11. Benichou O, Laredo J, Vernejoul M. Type II autosomal dominant osteopetrosis (Albers-Schönberg disease): clinical and radiological manifestation in 42 patients. Bone 2000; 26: 87-93. [ Links ]
12. Vernejoul M. Sclerosing bone disorders. Best Pract Res Clin Rheum 2008; 22(1): 71-83. [ Links ]
13. Toro C, Quintana M, Restrepo J, Rondón F, Cons F, Iglesias A, et al. Osteoesclerosis axiales. Propuesta para una nueva aproximación diagnóstica. Rev Colomb Reumatol 2004; 11(4): 341-346. [ Links ]
14. Waguespack S, Hui S, DiMeglio L, Econs M. Autosomal Dominant Osteopetrosis: Clinical Severity and Natural History of 94 Subjects with a Chloride Channel 7 Gene Mutation. The J Clin Endocrinol Metab 2007; 92(3): 771-778. [ Links ]
15. Gelb B, Shi G, Champan H, Desnick R. Pycnodysostosis, a lysosomal disease caused by cathepsin K deficiency. Science 1996; 273: 1236-1238. [ Links ]
16. Iglesias-Gamarra A, Vázquez-Lamadrid J, Abud C. Enfermedades metabólicas del hueso. Bogotá - Colombia, Instituto Nacional de Salud 1992 volumen II: p. 608-613. [ Links ]
17. Marreño L, Rondón V, Barbán D, Morales E, Quintana F. Estudio en una familia de una paciente con picnodisostosis. Rev Cubana Ortop Traumatol 2004; 18(1): 34-40. [ Links ]
18. Russel G, Mueller G, Shipman C, Croucher P. Clinical discords in bone resortion. Novartis Found Symp 2001; 232: 251-257. [ Links ]
19. Gazzerro E, Canalis E. Bone morphogenetic proteins and their antagonists. Rev Endocr Metab Disord 2006; 7: 51-65. [ Links ]
20. Caputo S, Couprie J, Duband-Goulet I, Konde E, Lin F, Braund S, et al. The Carboxyl-terminal Nucleoplasmic Region of MAN1 Exhibits a DNA Binding Winged Helix Domain. The J Biol Chem 2006; 281(26): 18208-18215. [ Links ]
21. Holmer L, Worman H. Inner nuclear membrane proteins: functions and targeting. CMLS Cell Mol Life SCi 2001; 1741-1747. [ Links ]
22. Li Y, Xiao Z. Advances in Runx2 regulation and its isoforms. Med Hypotheses 2007; 68: 169-175. [ Links ]
23. Valcourt U, Moustakas A. BMP signaling in Osteogenesis, Bone Remodeling and Repair. Europ J Trauma 2005; 5: 464-479. [ Links ]
24. Elke P, Boudin E, Van Hul W. Wnt signaling: A Win For bone. Arch Biochem and Bioph 2008; 473: 112-116. [ Links ]
25. Liu F, Kohlmeier S, Wang C. Wnt signaling and skeletal development. Cell Signal 2008; 20: 999-1009. [ Links ]
26. Zimmermann B. Effects of pyrophosphate on desmal and endocondral mineralization and TNAP activity in organoid culture. Ann Anat 2008; 167-177. [ Links ]
27. Gurley K, Reimer R, Kingsley D. Biochemical and Genetic Analysis of ANK in Arthritis and Bone Disease. The Am J Hum Gen 2006; 79: 1017-1029. [ Links ]
28. Balemans W, Patel N, Ebeling M, Van Hul E, Wuyts W, Lacza C, et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet 2002; 39: 91-97. [ Links ]
29. Vanhoenacker F, Balemans W, Tan G, Dikkers F, De Schepper A, Mathysen D, et al. Van Buchem disease: lifetime evolution of radioclinical feactures. Skeletal Radiol 2003; 32: 708-718. [ Links ]
30. Balemans W, Van Den Ende J, Freire A, Dikkers F, Willems P, Vanhoenacker F, et al. Localization of the gene for Sclerosteosis to the van Buchem disease-Generegion on chromosome 17q12-q21. Am J Hum Genet 1999; 64: 1661-1669. [ Links ]
31. Boiden L, Mao J, Belsky J, Mitzner L, Farhi A, Mitnick M, et al. High bone density due to mutation in LDLreceptor- relat protein 5. N Engl J Med 2002; 346: 1513-1521. [ Links ]
32. Johnson M, Gong G, Kimberling W, Recker S, Kimmel D, Recker R. Linkage of a gene causing high bone mass to human chromosome 11(11q12-13). Am J Hum Genet 1997; 60: 1326-1332. [ Links ]
33. Balemans W, Piters E, Cleiren E, Ai M, Van Wesenbeeck L, Warman M, et al. The Binding Between Sclerostatin and LRP5 in altered by Dkk1 and by High-Bone Mass LRP5 mutations. Calcif Tissue Int 2008 in press. [ Links ]
34. Millar D, Maisels D, Batstone J, Yates B. Craneofacial Surgery in Craniometaphyseal dysplasia. Am J Surg 1967; 113: 615-621. [ Links ]
35. Reichenberger E, Tiziani V, Watanabe S, Park L, Ueki Y, Santanna C, et al. Autosomal dominant craniometaphyseal dysplasia is caused by mutations in the transmembrane protein ANK. Am J Hum Genet 2001; 1321-1326. [ Links ]
36. Day R, Park T, Ojemann J, Kaufman B. Foramen magnum descompresssion for cervicomedulary encroachment in craniometaphyseal dysplasia: Case report. Neurosurgery 1997; 41: 960-964. [ Links ]
37. Sheppard W, Shprintzen R, Tatums, Woods C. Craniometaphyseal dysplasia: a case report and review of medical and surgical management. Int J Pediatr Otorhinolaryngo 2003; 67: 71-77. [ Links ]
38. Lin F, Blake D, Callebaut I, Skerjanc I, Holmer L, McBurney M, et al. MAN1, an inner nuclear membrane protein that shares the LEM domain with laminaassociated polypeptide 2 and emerin. J Biol Chem 2000; 275: 4840-4847. [ Links ]
39. Couto A, Bruges-Armas J, Peach C, Chapman K, Brown M, Wordsworth B, et al. A Novel LEMD3 Mutation Common to Patients with Osteopoiquilosis With and Without Melorheostosis. Calcif Tissue Int 2007; 81: 81-84. [ Links ]
40. Ariza A, Egea E, Loeza F, Barrera C, Donato M, Iglesias A, et al. El pleomorfismo clínico de la escleroderma lineal. Acta Med Col 1989; 14: 71-81. [ Links ]
41. Menten B, Buysse K, Zahir F, Hellemans J, Hamilton S, Costa T, et al. Osteopoiquilosis, short stature and mental retardation as key feactures of a new microdelection syndrome on 12q14. J Med Genet 2007; 44: 264-268. [ Links ]
42. Chaudier-Mnaymneh L, Broder, Mnaymneh W. Lobular carcinoma of the breast metastasi to bone with usual clinical Cancer 1984; 52: 1801-1803. [ Links ]
43. Takayanagy H. Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. Nat Rev Immunol 2007; 7: 292-304. [ Links ]
44. Data H, Ng W, Walker J, Tuck S, and Varanasi S. The cell biology of bone metabolism. J Clin Pathol 2008; 61: 577-587. [ Links ]
45. Iba K, Takada J, Kamasaki H, Oda T, Hatakeyama N, Wada T, et al. A significant improvement in lower limb pain after treatment with alendronate in two cases of Camurati-Engelmann disease. J Bone Miner Metab 2008; 26: 107-109. [ Links ]
46. Bondestam J, Mäyränpää M, Ikegawa S, Marttinen E, Kröger H, Mäkitie O. Bone biopsy and densitometry findings in a child with Camurati-Engelmann disease. Clin Rheumatol 2007; 26: 1773-1777. [ Links ]
47. Janssen K, Gershoni-Baruch R, Van Hul E, Brik R, Guañabens N, Migone N, et al. Localisation of the gene causing diaphyseal dysplasia Camurati- Engelmann to chromosome 19q13. J Med Genet 2000; 37: 245-249. [ Links ]
48. Iglesias-Gamarra A, Restrepo JF, Lacouture M, Iglesias- Rodríguez A, Calvo E, Rondon F. Distrofia ósea mixta no esclerosante. Paquidermoperiostosis y displasia diafisiaria tipo Engelmann-Camurati. Rev española de enf. metabólicas óseas. 2000; 9(5): 178- 183. [ Links ]
49. Hernández M, Peris P, Guañabens N, Alvarez L, Monegal A, Pons F, et al. Biochemical Markers of Bone Turnover in Camurati-Engelmann Disease: A Report on Four Cases in One Family. Calcif Tissue Int 1997; 61: 48-51. [ Links ]
50. Janssens K, Vanhoenacker F, Bonduelle. Camurati- Engelmann Disease: revive of the clinical, radiological and molecular data of 24 families and implications towards diagnostics and treatment. J Med Genet 2006; 43: 1-11. [ Links ]
51. Van Wesenbeek L, Cleiren E, Gram J, Beals R, Bénichu O, Scopellini D, et al. Six novel missense mutations in the LDL receptor-related protein 5 (LRP5) gene in different conditions with an increased bone density. Am J Hum Genet 2003; 72: 763-771. [ Links ]
52. Hernandez-Cassis C, Vogel C, Hernandez T, Econs M, Iglesias M, Iglesias A, et al. Autosomal Dominant Hyperostosis/Osteosclerosis with High Serum Alkaline Phosphatase Activity. J Clin Endocrinol Metab 2003; 88(6): 2650-2655. [ Links ]

