










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Colombiana de Reumatología
versión impresa ISSN 0121-8123
Rev.Colomb.Reumatol. v.16 n.1 Bogotá ene./mar. 2009
ARTÍCULO DE REVISIÓN
Behcet's disease
Ana Milena Toro Giraldo1, Luis Fernando Pinto Peñaranda2,
Carlos Jaime Velásquez Franco2, Javier Darío Márquez Hernández2
1 Dermatóloga - Docente de dermatología, Universidad Pontificia Bolivariana, Medellín, Colombia.
2 Reumatólogo del Hospital Pablo Tobón Uribe y Profesor Asociado de la Universidad Pontificia Bolivariana, Medellín, Colombia.
Recibido: Noviembre 15/2008 Aceptado: Febrero 5/2009
La enfermedad de Behcet (EB) es una enfermedad multisistémica de causa desconocida, caracterizada por un curso crónico, recurrente y compromiso inflamatorio de los vasos de todos los calibres. Los principales hallazgos clínicos incluyen: aftas orales y genitales, artritis, lesiones cutáneas, manifestaciones oculares, gastrointestinales y neurológicas1-7.
Historia4
La primera descripción de la sintomatología de la enfermedad fue publicada por Hipócrates, en el siglo V antes de Cristo, en su tercer libro de epidemiología8:
"There were other forms of fever... Many developed aphtae, ulcerations. Many ulcerations about the genital parts... Watery ophtalmies of a chronic character, with pains; fungus excretions of the eyelids externally, internally, which destroyed the sight of many persons... there were fungous growths on ulcers, and on those localized on the genital organs. Many anthraxes through the summer... other great affections: many large herpetes".
A principios del siglo XX, Blüthe, Planner, Remenovsky y Shigeta reconocieron la tríada de iritis, úlceras mucocutáneas y genitales.
En 1930, Adamantiades presentó a la sociedad médica de Atenas el caso de un paciente de 20 años de edad quien desarrolló iritis recurrente con pérdida de la visión, flebitis, ulceraciones genitales, orales y artritis en la rodilla, el cual fue publicado un año después.
En 1924, el dermatólogo turco Hulusi Behçet examinó un paciente con historia de estomatitis aftosa recurrente, úlceras genitales, eritema nodoso y afección ocular; en los siguientes 12 años evaluó varios casos similares, los cuales fueron publicados en 1937 y a partir de entonces este síndrome fue reconocido con el nombre de enfermedad de Behcet.
Epidemiología
La EB tiene una distribución mundial; sin embargo, su prevalencia varía mucho según la ubicación geográfica (Tabla 1), siendo mayor en los países correspondientes con la antigua ruta de la seda y menor en el norte de Europa, Estados Unidos e Inglaterra1,5,7.
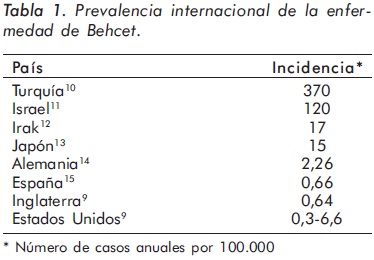
La edad promedio de inicio de los síntomas varía entre los 20 y 40 años y rara vez inicia en niños y en ancianos1,5. La prevalencia por géneros es variable; en el medio oriente predomina en hombres, con una relación hombre / mujer de 3,8: 1 en Israel, 5,3: 1 en Egipto y 3,4: 1 en Turquía. En Alemania, Japón y Brasil existe un ligero predominio femenino, y en Estados Unidos la relación mujer / hombre es de 5:1.
La única serie publicada en Colombia muestra un predominio del género femenino (8,7:1), una edad al diagnóstico de 45 años (36-52,5) y un año de retardo entre el primer síntoma y el diagnóstico de EB16.
La gravedad de la enfermedad es mayor en los hombres9,17, quienes tienen mayor probabilidad de desarrollar aneurismas, afección ocular, tromboflebitis y enfermedad neurológica; mientras que en las mujeres es más frecuente el eritema nodoso9.
La EB es poco común en pacientes de raza negra y en ellos predomina más el compromiso mucocutáneo18.
Patogénesis
La patogénesis de la EB es desconocida. Se ha relacionado con la presencia de anormalidades inmunológicas, posiblemente inducidas por agentes infecciosos o ambientales en individuos genéticamente susceptibles. Hallazgos recientes han soportado la importancia de los factores genéticos y han permitido definir mejor la naturaleza de la inflamación en esta entidad19.
Susceptibilidad genética
La EB no se hereda con un modelo de transmisión mendeliano. La mayoría de los casos son esporádicos. Sin embargo, se han identificado varios casos de agregación familiar con un riesgo elevado de enfermedad entre los familiares de primer grado19-22. El análisis de dichos casos ha soportado la existencia de un complejo modelo de transmisión, con anticipación genética (enfermedad de inicio más temprano) en las generaciones sucesivas23,24. Asimismo, se ha encontrado que la prevalencia de HLA-B51 es mucho más alta que en los casos esporádicos20.
La asociación de la EB con la presencia de HLA-B51 constituye la evidencia más fuerte que involucra los factores genéticos en su patogénesis. Ha sido largamente investigado si el HLA-B51 desempeña un papel directo en su patogénesis o si esta asociación refleja un desequilibrio de unión con un gen de susceptibilidad para la EB localizado cerca al locus del HLA-B. El gen del TNF y el gen de la linfotoxina, los cuales están localizados centroméricos al HLA-B, han sido el blanco de atención como posibles candidatos para susceptibilidad a la enfermedad de Behcet19.
La investigación del segmento genómico entre el TNF y el locus del HLA-B reveló una asociación más fuerte de la EB con el gen MICA (MHC class I chain related gene A); este gen es expresado en fibroblastos, células epiteliales, células endoteliales y monocitos. Se propuso inicialmente que este patrón de expresión podía explicar los sitios de inflamación y que la presentación antigénica por las moléculas MICA a las células T, la cual está aumentada en la EB, pudiera ser un mecanismo patogénico importante. Sin embargo, se ha demostrado que los polimorfismos de la molécula MICA no tienen un papel en la presentación antigénica a las células T en el epitelio intestinal y que el antígeno MICA no parece estar expresado en la membrana celular de los queratinocitos y monocitos, a pesar de la detección de RNAm19,25.
Estudios de mapeo usando microsatélites altamente polimórficos confirmaron la región crítica para la EB como el segmento de 46kb entre el locus MICA y el HLA-B26. Estudios de asociación alélica, diferenciación genotípica y análisis de estratificación en diferentes grupos étnicos han probado que el HLA-B51 es el que muestra más fuerte asociación con la EB y todas las otras asociaciones, incluyendo el MICA, son debidas a un desequilibrio de unión con el HLA-B5113,27,28. Sin embargo, este desequilibrio de unión puede extenderse distancias muy largas dentro de la región del MICA y es aún difícil valorar los efectos suplementarios del gen MICA o los otros genes vecinos en la susceptibilidad a la enfermedad19.
Diferentes hipótesis han surgido para explicar el papel directo patogénico del HLA-B51 en la EB19:
-
El HLA-B51 es uno de los antígenos divididos del HLA-B5 y difiere sólo por dos aminoácidos de otros antígenos divididos, los cuales no están asociados con la EB.
-
Participación en la presentación de péptidos antigénicos microbianos relacionados con la EB.
-
Reactividad cruzada entre el HLA-B51 y antígenos específicos de diferentes órganos.
Veinticuatro diferentes alelos de HLA-B51 (HLA-B* 5101 - HLA-B*5124) han sido descritos; todos ellos comparten la misma secuencia de aminoácidos en el bolsillo B de la ranura ligadora de antígeno excepto B*5107 y B*5120. Tipificación molecular de HLA-B51 en diferentes grupos étnicos sugiere que los alelos B51 en los pacientes con EB no son diferentes de aquellos de los controles sanos7,19,29.
Otra función de las moléculas de HLA clase I ha sido recientemente descrita por la identificación de una nueva familia de receptores expresados principalmente por las células NK, CD8+ y menos frecuentemente por las células CD4+, conocidos como receptores KIR (killer immunoglobulin-like receptors). La interacción con estos receptores se asocia con inhibición selectiva de las células NK o de la citotoxicidad mediada por células. Una hipótesis alternativa es que el papel patogénico del HLA - B51 en la EB podría resultar de una interacción con moléculas KIR en las células inflamatorias1,19.
En un estudio reciente se demostró que la contribución del locus del HLA-B a la susceptibilidad genética global de la EB es menor del 20%30 y se espera la identificación de otros locus que confieran mayor susceptibilidad genética a padecer esta enfermedad19.
Finalmente, pueden contribuir a la susceptibilidad y/o severidad en la enfermedad los polimorfismos en genes que codifican otras moléculas efectoras, tales como: proteínas TAP, moléculas de adhesión intercelular (ICAM-1), interleuquinas, quimoquinas y receptores de quimoquinas (CCL2 /MCP-1), lectina ligadora de manosa (MLB), N-acetil transferasa (NAT2), sintasa de óxido nítrico endotelial (eNOS), glutatión S-transferasa (GST), citocromo P450 (CYP1A1), HSPs y FcγR. El papel de los polimorfismos y las mutaciones protrombóticas (factor V Leiden) es controvertido.20
Agentes microbianos
Éstos han sido postulados como agentes causales o factores disparadores de la enfermedad; sin embargo, su papel permanece incierto y la asociación con la EB ha sido sugerida por la mayor prevalencia de ésta en condiciones de hacinamiento, grupos socioeconómicos bajos y grupos familiares31. Los hallazgos son controvertidos: algunos investigadores han reportado el aislamiento de algunos virus de las lesiones mucocutáneas; no obstante, esto no ha podido ser confirmado por otros1,19. Argumentos a favor de la asociación del HSV con la EB incluyen la identificación de genoma del HSV tipo 1 en linfocitos circulantes, saliva, úlceras genitales y gastrointestinales de pacientes con EB y la descripción de un modelo experimental en ratones inoculados con HSV, en el cual el 30% desarrolló manifestaciones parecidas a EB1,4,19,32,33.
Un estudio reciente muestra evidencia que sugiere un papel causal de parvovirus B19 en la EB, sugiriendo la presencia del virus, particularmente en lesiones cutáneas no ulcerativas34.
Algunas bacterias también han sido involucradas en la patogénesis de la EB; los argumentos a favor de esta teoría incluyen: una elevada reactividad a la inyección cutánea de antígenos estreptocócicos, logrando inducir manifestaciones sistémicas de la EB en algunos pacientes; la aparición de las manifestaciones clínicas después de procedimientos de extracción dental; la mayor prevalencia de caries dental, periodontitis o tonsilitis; el elevado porcentaje de colonias de Streptococcus sanguis (S. sanguis) en la flora oral y la detección de anticuerpos reactivos contra serotipos poco comunes de S. sanguis en el suero de dichos pacientes1,4,35,36
Las proteínas de choque térmico (HSP) han sido propuestas como el común denominador entre diferentes agentes microbianos, por presentar homología estructural con HSP mitocondriales humanas. Ciertos epítopes de HSP microbianas actuarían como disparadores de una respuesta inmune específica que produciría una reacción inflamatoria cruzada, a través de un mecanismo de mimetismo molecular, que llevaría a una activación de células T y de células de memoria y que determinaría la cronicidad y la naturaleza recurrente y remitente de las lesiones de la EB1,37.
Mecanismos inmunes
El principal hallazgo microscópico de la EB activa es una oclusión vascular inflamatoria (trombo inflamatorio); en los infiltrados perivasculares se encuentran células T CD4+ y células Th1 que responden a varios estímulos, con producción de IL-2, IFN-γ y TNF-β38. Adicionalmente, se han encontrado niveles elevados de IL-8 en pacientes con enfermedad activa39 y de IL-1β y factor de crecimiento fibroblástico en EB grave4. Otros autores demostraron elevación persistente de los niveles plasmáticos de IL-10, independiente de la actividad de la enfermedad, mientras que los niveles de IL-12 y del receptor soluble de TNF sólo se elevaron en la enfermedad activa; plantean que estos hallazgos servirían como marcadores biológicos de actividad de la enfermedad40.
Varios autores han demostrado una elevada proporción de células T γδ en lesiones de EB; estas células actúan como primera línea de defensa, controlan y mantienen el crecimiento e integridad epiteliales, reconocen estructuras microbianas, previniendo el paso de patógenos mediante citotoxicidad contra las células infectadas. Asimismo, un porcentaje alto de estas células podría producir citoquinas inflamatorias como IFNγ y TNFα1,41.
Se ha considerado también un incremento en la expresión de CD11a y CD18 en los neutrófilos, lo cual podría explicar su acumulación en los sitios inflamatorios4. De igual manera, ha sido recientemente demostrado que los neutrófilos de los pacientes con EB expresan constitutivamente RNAm del TNF y producen abundante cantidad de esta citoquina; esta producción aumentada de TNF podría activar los neutrófilos y perpetuar su ciclo de vida, resultando también en una acumulación de neutrófilos activados en el sitio de inflamación19. Por otro lado, la activación de los monocitos puede explicar la producción de citoquinas proinflamatorias responsables de la cronicidad de la inflamación4,42.
Un hallazgo bien conocido de la EB es la tendencia trombótica con predominio venoso. Varios autores han encontrado hallazgos consistentes con activación del sistema de coagulación y fibrinolisis, reflejando activación o injuria endoteliales19,43. Se ha sugerido que la activación endotelial por los infiltrados perivasculares de células mononucleares y neutrófilos explicaría el estado protrombótico de la EB. Sin embargo, los marcadores de activación endotelial no difieren entre los pacientes con o sin trombosis19.
La presencia de mutaciones procoagulantes tales como el factor V de Leiden o del gen de la protrombina incrementa el riesgo de trombosis en EB44,45. La contribución de los anticuerpos anticardiolipinas y anticélulas endoteliales a la injuria endotelial y a la tendencia trombótica de la EB es especulativa; estos anticuerpos podrían ser causantes de trombosis o ser secundarios a la injuria tisular19,46.
Tomando en consideración las evidencias anteriormente mencionadas, se ha propuesto un modelo que explica la patogénesis de la EB: un factor exógeno (virus o bacteria) es presentado por macrófagos y reconocido por células T CD4+ en el contexto de los antígenos del MHC clase II. Las células T Th1 activadas producen citoquinas (IL-2, IFN-γ , TNF-ß) e inducen una proliferación de las células B. El IFN-γ activa los macrófagos, que a su vez liberan TNF-α, IL-1 e IL-8, las cuales inducen la expresión de moléculas de adhesión en las células endoteliales; IL-8 también induce quimiotaxis y activación de neutrófilos; ambos eventos son responsables del paso de neutrófilos polimorfonucleares y linfocitos T activados a través del endotelio a la zona de inflamación4,38. Los factores genéticos pueden contribuir a la expresión y perpetuación de la enfermedad4.
Manifestaciones clínicas
La enfermedad de Behcet es un desorden multisistémico que puede afectar cualquier órgano. El curso clínico está caracterizado por exacerbaciones y remisiones de duración, frecuencia y pronóstico impredecibles; pueden existir variaciones de acuerdo a la herencia, el género, el medio ambiente y el estado socioeconómico1.
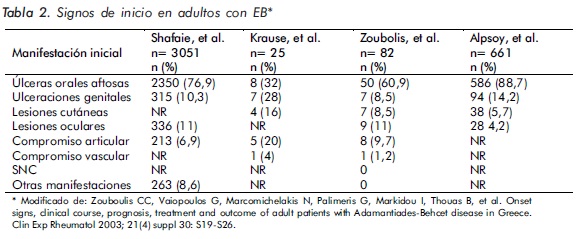
Varios estudios han analizado los signos de inicio más frecuentes en la EB16,47 (Tabla 2).
De igual manera, han sido analizados los hallazgos clínicos durante el curso de la EB16,47,48 (Tabla 3).
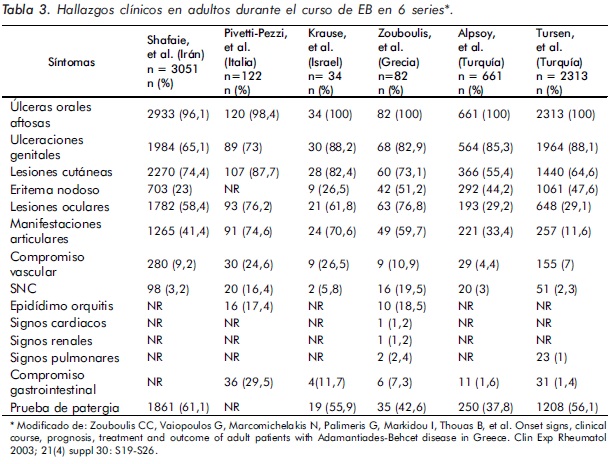
Las aftas orales se presentan casi en el 100% de los pacientes, por lo cual constituyen uno de los criterios mayores para el diagnóstico de la EB, siendo el hallazgo inicial en el 66% a 76% de los pacientes. Para ser consideradas manifestación de esta enfermedad deben recurrir al menos tres veces en un periodo de un año49.
Los sitios más afectados son: la mucosa bucal, la lengua, los labios, las encías, menos frecuentemente el paladar, la faringe posterior, la laringe o las amígdalas. Se inician como áreas elevadas eritematosas que, en uno a dos días, se convierten en úlceras redondas u ovales poco profundas con un discreto borde eritematoso, cubiertas en el fondo con una pseudomembrana blanca o amarilla. Las úlceras pequeñas pueden coalescer formando una úlcera grande y única, o pueden agruparse produciendo la denominada ulceración herpetiforme. Estas lesiones son usualmente dolorosas, curan sin dejar cicatriz, y su resolución ocurre en una a dos semanas. Los intervalos entre las recurrencias son variables y algunas veces se presentan en forma continua1,2,4,5,7.
Las aftas o úlceras genitales se pueden localizar en el escroto, el pene, la región perianal, la vagina y la vulva. Clínicamente, son similares a las orales, pero en general, son más grandes, más profundas, más dolorosas y toman más tiempo para curar que las lesiones orales; cuando se resuelven, dejan una cicatriz o decoloración blanca.
Raramente se presentan al inicio de la enfermedad y son menos recurrentes que las úlceras orales. En las mujeres, estas úlceras pueden relacionarse cronológicamente con el ciclo menstrual1,4,5.
Las úlceras orales y genitales recurrentes pueden ser la única manifestación de la enfermedad en mujeres jóvenes, incluso por varios años, antes de presentar otras manifestaciones de la EB.
La frecuencia de las manifestaciones cutáneas varía entre el 33 y el 100% de los casos; pueden presentarse, en el mismo paciente, varios tipos de lesiones, en forma simultánea o en diferentes ocasiones y hacen parte de los criterios diagnósticos de la EB50. Su incidencia varía ampliamente con la localización geográfica; algunos autores correlacionan algunas lesiones cutáneas específicas de la enfermedad con la edad de inicio, el género y la tipificación del HLA51.
Las manifestaciones cutáneas específicas incluyen: lesiones papulopustulares, lesiones similares al eritema nodoso, tromboflebitis de los vasos superficiales e hipersensibilidad cutánea (fenómeno de patergia) 52.
Las lesiones papulopustulares incluyen: pseudofoliculitis estériles o lesiones acneiformes en una base eritematosa; se manifiestan inicialmente como pápulas que evolucionan en 24 a 48 horas a pústulas. Se pueden localizar en cualquier parte del cuerpo, siendo más frecuentes en los miembros inferiores. Se debe hacer diagnóstico diferencial con acné en adolescentes o en personas en tratamiento con corticoides.
El eritema nodoso es más frecuente en las mujeres, afectando principalmente los miembros inferiores, típicamente la región pretibial; se caracteriza por nódulos eritematosos dolorosos, rodeados por un halo periférico; no se ulceran y se resuelven espontáneamente, dejando áreas de hiperpigmentación. Debe diferenciarse de la tromboflebitis superficial, que aparece en el 30% de los pacientes con EB.
Otros hallazgos cutáneos descritos con menor frecuencia incluyen: vesículas, pioderma gangrenoso, síndrome de Sweet, lesiones papulonodulares purpúricas acrales y raramente necrosis52-55.
Una característica importante en la EB es un incremento en la respuesta inflamatoria no específica19. El ejemplo clásico es la reacción cutánea de patergia, la cual se caracteriza por el desarrollo de una pápula o una pústula 24 a 48 horas después de una punción en la piel con aguja estéril, similar a la que aparece espontáneamente en la enfermedad. Este incremento en la respuesta al trauma menor no es solo en la piel y puede ser observado en otros sitios corporales o presentarse como un infiltrado inflamatorio perivascular, incluso sin manifestación cutánea ("patergia histológica")1,2,4,56.
La interacción entre moléculas de adhesión celular y la proliferación endotelial puede jugar un papel importante en la patogénesis del fenómeno de patergia56.
Existe variación geográfica en la frecuencia del fenómeno de patergia, siendo usualmente positivo en los pacientes con EB que habitan las regiones correspondientes a la antigua ruta de la seda, pero sólo en el 20% a 30% de los americanos y los europeos1,2.
El fenómeno de patergia es usualmente positivo durante la fase activa de la EB y negativo o débilmente positivo cuando la enfermedad se encuentra en remisión1,2. La intensidad de la reacción puede variar en el mismo paciente en diferentes momentos, y usualmente existe variación intra e interobservador. Muchos factores pueden influenciar la interpretación de la prueba de patergia, tales como: el tipo de aguja, el retraso en la lectura y la técnica de punción empleada57; todos estos factores, sumados a la falta de estandarización, ocasionan la gran diferencia en los resultados en los distintos grupos poblacionales estudiados2,58.
Las manifestaciones oculares se presentan en cerca del 70% de pacientes con EB59,60, razón por la cual se considera un criterio de diagnóstico de la entidad. Se pueden afectar casi todas las estructuras oculares, de forma uni o bilateral. Son más frecuentes en los hombres, con una relación de 2:1. Característicamente, los episodios son recurrentes, produciendo daño ocular irreversible. La edad promedio de inicio es alrededor de los 30 años. En los hombres, se caracteriza por tener un inicio más temprano y un curso más grave59. Los factores de riesgo que se correlacionan con el compromiso ocular en la EB son: la edad de comienzo de la enfermedad, el género masculino y compromiso del sistema nervioso central (SNC)1.
La típica afección ocular es la uveítis recurrente, que puede ser anterior, posterior o panuveítis. La uveítis anterior o iridociclitis puede estar o no acompañada de hipopion. Otros hallazgos incluyen conjuntivitis, epiescleritis, escleritis y queratitis. Es poco frecuente encontrar parálisis de los músculos extraoculares por compromiso neurológico1-4,59,60.
La afección del segmento posterior es más frecuente y grave y se acompaña siempre de vitriítis; las alteraciones pueden ser de tres tipos59:
- Vasculitis retiniana, que afecta sobre todo las venas (periflebitis) y puede dar lugar a oclusión venosa.
- Exudación retiniana aguda masiva, que afecta las capas retinianas exteriores y a veces da lugar a grandes áreas de atrofia retiniana por obliteración de los vasos sanguíneos situados por encima de ésta.
- Vasodilatación retiniana. Es el hallazgo más frecuente y duradero; el consiguiente aumento de la permeabilidad vascular provoca edema retiniano difuso, edema macular cistoideo e hiperemia del disco óptico.
La oclusión vascular retiniana ha sido correlacionada con tres factores principales: estasis vascular, anormalidades trombóticas y vasculitis1.
La pérdida de la visión es la más importante y temida complicación de la EB; ocurre, en promedio, 3,36 años después del comienzo de los síntomas oculares1. En algunas series se informa que hasta el 74% de los pacientes tratados perdieron la visión útil entre seis y diez años después del comienzo de los síntomas oculares61.
Sakamoto et al. demuestran que la presencia de lesiones cutáneas, artritis y el compromiso ocular posterior se correlacionan con un mal pronóstico visual; por el contrario, en el género femenino, la presentación cíclica de la enfermedad y el compromiso ocular anterior se asocian a mejor pronóstico62.
La incidencia de compromiso vascular varía entre el 7% y el 29%63; la EB afecta tanto el sistema arterial como el venoso. Se han descrito cuatro tipos de lesiones que incluyen: oclusión arterial, aneurismas, oclusión venosa y tromboflebitis o trombosis en las extremidades y en otros órganos. Las lesiones vasculares usualmente ocurren en el sistema venoso (80%), seguido por el sistema arterial (20%). La frecuencia de síntomas vasculares en los pacientes es de un 8,7% en hombres y de un 6,3% en mujeres64. El espectro de lesiones descritas incluye desde tromboflebitis superficial a lesiones oclusivas de la vena cava, estenosis y aneurismas del arco aórtico, la aorta torácica y abdominal, las arterias pulmonares y las periféricas. Aunque la complicación vascular más frecuente es la trombosis venosa, la principal causa de muerte es la ruptura de aneurismas de las grandes arterias, con una mortalidad que puede llegar a ser del 20%. Las lesiones oclusivas usualmente ocurren en grandes arterias, tales como la subclavia, la pulmonar y las femoropoplíteas65; pero también puede comprometer arterias más pequeñas. Ha sido descrito infarto agudo de miocardio por trombosis y vasculitis coronaria66,64.
La frecuencia de manifestaciones neurológicas varía entre el 5% y el 30%, y es más común en los hombres. Tanto el SNC como el periférico pueden estar comprometidos. Los hallazgos en el SNC pueden ser divididos en dos grupos principales: el primero conformado por compromiso parenquimatoso, el cual incluye afección de tallo cerebral, manifestaciones hemisféricas, lesiones del cordón espinal y cuadros de meningoencefalitis. El segundo grupo corresponde al compromiso no parenquimatoso; éste incluye: trombosis del seno dural, oclusión y/o aneurismas arteriales. Las neuropatías periféricas y las miopatías son relativamente raras67.
Akman-Demir, et al., en 200 pacientes con neuro-Behcet, encontraron que el 81% presentó compromiso parenquimatoso (tallo cerebral 51%, cordón espinal 14%, compromiso hemisférico 15% y signos piramidales aislados 19%), mientras que el 19% presentó compromiso no parenquimatoso. En el primer grupo, los hallazgos más comunes fueron: signos piramidales, hemiparesia, cambios de conducta y disturbios esfinterianos; por otro lado, en el segundo grupo, el síndrome de hipertensión intracraneana debido a trombosis del seno dural fue la principal manifestación clínica. En el 60% de los casos con compromiso parenquimatoso el estudio del líquido cefalorraquídeo (LCR) mostró pleocitosis y/o hiperproteinorraquia, y en más de la mitad de pacientes de este grupo la resonancia magnética mostró lesiones del tallo cerebral o de los ganglios basales. Asimismo, se encontró que la presencia de compromiso parenquimatoso, elevación de proteínas o pleocitosis en el LCR, enfermedad de curso progresivo primario o secundario y recaídas durante el desmonte de esteroides, se asociaron con un peor pronóstico68.
Entre el 50% y el 60% de los pacientes presentan artralgias o artritis; ésta no es migratoria y ni erosiva, con remisiones y recaídas prolongadas1; las recaídas usualmente se asocian con fiebre, lesiones cutáneas (principalmente papulopustulares)69 y elevación de reactantes de fase aguda70. Típicamente la artritis es oligoarticular en la mayoría de los pacientes; la articulación más afectada es la rodilla, seguida por los tobillos, los codos y las muñecas; aunque también pueden comprometerse las pequeñas articulaciones de los pies y las manos.
Otros hallazgos, reportados con menor frecuencia, son: osteonecrosis con múltiples lesiones osteolíticas reversibles, entesitis, sacroiliítis, daño erosivo con pérdida del cartílago y formación de pannus1,4,5,71.
En el grupo de manifestaciones menos frecuentes se incluyen las cardiopulmonares, gastrointestinales y genitourinarias.
La prevalencia real del compromiso pulmonar en la EB es desconocida. En un análisis retrospectivo de 2.179 pacientes con EB, la prevalencia fue del 1,1%. La arteritis con formación de aneurismas es la principal manifestación de la EB; también se describen: oclusión trombótica de los vasos, infarto o hemorragia pulmonares, hipertensión arterial pulmonar y falla cardiaca derecha72.
La frecuencia de las manifestaciones cardíacas es del 5% al 10%4; éstas incluyen: infarto del miocardio, pericarditis, endocarditis, y anormalidades valvulares tales como regurgitación aórtica y mitral. La fibrosis endomiocárdica de los ventrículos puede ser secuela de la vasculitis y se puede complicar con trombosis intraventricular4,5.
En cuanto al compromiso gastrointestinal, se pueden encontrar úlceras únicas o múltiples a lo largo de todo el tracto gastrointestinal, siendo más frecuentes en el ileon terminal y en el ciego. Los principales hallazgos son dolor abdominal, diarrea, hemorragia y perforación4,5. Raramente se ha descrito el síndrome de Budd-Chiari que es un hallazgo de mal pronóstico73.
La incidencia de epididimitis es de 4% a 11%. Otros hallazgos menos frecuentes incluyen: glomerulonefritis aguda, amiloidosis, nefropatía por IgA, trombosis de la vena renal y glomerulonefritis focal o segmentaria4.
En Colombia, Londoño y colaboradores encuentran aftas orales en el 100%, aftas genitales en el 75% y algún tipo de compromiso ocular en el 87,5% de los pacientes16.
Diagnóstico
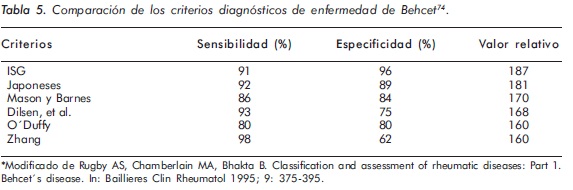
El diagnóstico se basa en hallazgos clínicos específicos y no en hallazgos de anatomía patológica o de laboratorio. Han sido propuestos diferentes criterios diagnósticos1,2,4; los del Grupo Internacional de Estudio50 (Tabla 4) tienen la mayor especificidad (96%) y el mayor valor relativo de discriminación74 (Tabla 5).

Los hallazgos histopatológicos no son específicos e incluyen: vasculitis linfocítica y necrotizante de las vénulas post capilares superficiales con necrosis fibrinoide asociada75-77. Chen et al. demostraron vasculitis casi en el 50% de los pacientes78. La inflamación endotelial es un hallazgo frecuente, así como un infiltrado linfocítico o neutrofílico perivascular asociado77. Hallazgos adicionales incluyen: un infiltrado neutrofílico dérmico difuso con o sin formación de abscesos, que corresponden clínicamente a lesiones pustulares, foliculitis aguda y cambios pustulares acneiformes. Las lesiones de eritema nodoso corresponden a una vasculitis necrotizante de los vasos subcutáneos, usualmente asociada con trombosis. También se ha descrito paniculitis lobular o septal. La tromboflebitis superficial está presente en más del 30% de los pacientes. Las aftas orales y genitales muestran ulceración no específica, acompañada, en algunas ocasiones, por vasculitis linfocítica o leucocitoclástica77.
Los hallazgos de laboratorio son inespecíficos y reflejan el estado inflamatorio de la enfermedad; éstos incluyen: elevación de la velocidad de eritrosedimentación, la proteína C reactiva y otros reactantes de fase aguda. Ocasionalmente puede haber elevación de los niveles de IgA, IgG, α-2 globulina, IgM y depósito de complejos inmunes en algunas lesiones4.
Debido a que existen pocos marcadores de laboratorio confiables que sirvan como parámetro de seguimiento de la EB, la evaluación de la actividad de la enfermedad se hace con base en las manifestaciones clínicas. Por la necesidad de tener un método objetivo para evaluar la actividad y progresión de la EB y su respuesta al tratamiento, se diseñó el Current Activity Form (BDCAF), basado en el Iranian Behçet's Disease Dynamic Measure (IBDDAM) y en un esquema europeo. Este método fue revisado y aprobado en una reunión de consenso del International Scientific Committee on Behcet Disease realizado en Leeds en 199479,80.
El BDCAF evalúa varios parámetros que incluyen: la escala de Likert (indica cómo perciben el médico y el paciente el comportamiento de la enfermedad en las últimas cuatro semanas) y la presencia o ausencia (en las cuatro últimas semanas) de aftas orales o genitales, compromiso articular, cutáneo, gastrointestinal, ocular, SNC y vascular. Este instrumento ha sido validado y tiene una buena confiabilidad interobservador en la valoración de la actividad general de la enfermedad; su uso ha sido sugerido en el monitoreo clínico periódico de los pacientes con EB y en la evaluación de resultados terapéuticos80.
Otra forma de evaluar la actividad de la enfermedad es a través de un índice de actividad clínica que evalúa los hallazgos clínicos en los últimos seis meses acorde con lo reportado por Yacizi, et al81.
Por otro lado, Krause, et al82. sugirieron una estimación de severidad de la enfermedad, clasificándola como leve, moderada o severa, según la presencia de hallazgos clínicos específicos.
Pronóstico
La EB tiene un curso crónico con exacerbaciones y remisiones impredecibles, cuya frecuencia y severidad pueden disminuir con el tiempo. Después de los primeros cinco años, la enfermedad adquiere un curso estable o hacia la mejoría; después de la cuarta década, se disminuye la severidad clínica, con intervalos más largos de recurrencia entre las exacerbaciones83. El pronóstico es bueno, a menos que haya compromiso de órganos vitales2,4.
La presentación en el género masculino y la edad de inicio temprano (antes de los 25 años) se han asociado con manifestaciones más graves de la enfermedad, que incluyen: trombosis vascular y compromiso ocular, gastrointestinal o del SNC84-86.
Kural-Seyahi86, et al. encontraron una tasa de mortalidad del 9,8%, debida principalmente al compromiso vascular. Asimismo, el compromiso neurológico y la perforación de úlceras intestinales también representan causas importantes de mortalidad86.
La positividad para el HLA-B51 generalmente no determina el pronóstico ni la respuesta a la terapia. En un estudio comparativo de Turquía y Reino Unido no se encontró asociación entre la presencia del alelo HLA-B51 y la gravedad de la enfermedad84.
Tratamiento
Las aftas orales y genitales se manejan con la aplicación tópica de corticosteroides de alta potencia, como terapia de primera línea. En las aftas menores o mayores graves están indicados los corticoides intralesionales (triamcinolona). El uso de corticoides sistémicos está reservado para lesiones mucocutáneas más graves y extensas5,87,88.
El sucralfate induce proliferación de los fibroblastos dérmicos y formación de tejido de granulación. Un estudio doble ciego demostró que el uso de sucralfate en suspensión redujo la frecuencia, el dolor y el tiempo de curación de las aftas orales y genitales. Los resultados de este estudio también sugieren que puede ser usado en la prevención del desarrollo de aftas orales en pacientes con EB. La dosis recomendada es 5 ml cuatro veces al día como suspensión oral y aplicación tópica en lesiones genitales89.
Para el manejo del dolor se puede utilizar lidocaína viscosa al 1% o 2% tópica.
Otras terapias tópicas incluyen clorhexidina 1% a 2%, tetraciclina en suspensión y tacrolimus88.
El medicamento sistémico de elección, en el tratamiento de las aftas orales y genitales, es la colchicina; su mecanismo de acción se basa en la inhibición de la quimiotaxis de los neutrófilos. La colchicina oral (0,6 mg tres veces al día) disminuye en más del 50% el tamaño, la frecuencia y la duración de las úlceras orales90. Es también de utilidad en el manejo de las lesiones cutáneas5.
Para casos más graves y refractarios está indicado el uso de dapsona que también inhibe la quimiotaxis de los neutrófilos91.
Algunos autores han reportado que la pentoxifilina disminuye la frecuencia y severidad de las aftas orales y vaginales a corto plazo en un 75%; este efecto continúa por más de seis meses en el 50% de los pacientes. El uso combinado de pentoxifilina y colchicina parece tener un efecto sinérgico en las úlceras orogenitales92,93.
La azatioprina es el inmunosupresor con mayor evidencia científica en la EB; un estudio prospectivo, doble ciego, controlado con placebo demostró reducción en la frecuencia y la severidad de las lesiones orales y cutáneas87.
Varios estudios han encontrado que la talidomida es efectiva en el manejo de las lesiones mucocutáneas, úlceras gastrointestinales y artritis87. El mecanismo de acción en la EB es a través de la modulación de la citotoxicidad mediada por complejos inmunes circulantes y por neutrófilos. Está reservada para enfermedad grave y refractaria a otros tratamientos, por sus potenciales efectos adversos, como neuropatía periférica y teratogenicidad5,87,94.
Las manifestaciones cutáneas, especialmente el eritema nodoso, responden satisfactoriamente a colchicina, dapsona o corticoides sistémicos4.
Lin et al., en 2006, recomiendan el uso de corticoides tópicos o intralesionales, pentoxifilina, sucralfate o dapsona, para enfermedad mucocutánea leve a moderada; dependiendo de la respuesta, estos tratamientos pueden ser usados solos o en combinación. Para controlar el dolor, es útil la lidocaína viscosa aplicada tópicamente. Si después de tres a cuatro semanas estas terapias son inefectivas, puede adicionarse colchicina oral y/o corticoides sistémicos a bajas dosis. Si en tres a cuatro semanas no hay mejoría, se debe considerar agregar azatioprina o metotrexate. Si fallan todas estas intervenciones, el siguiente paso es el uso de inhibidores del TNF-α, tales como infliximab o etanercept. El uso de ciclosporina e interferón α-2a solo está indicado cuando no haya respuesta con inhibidores del TNF-α87.
La Tabla 6 resume los esquemas de tratamiento indicados en el compromiso sistémico.
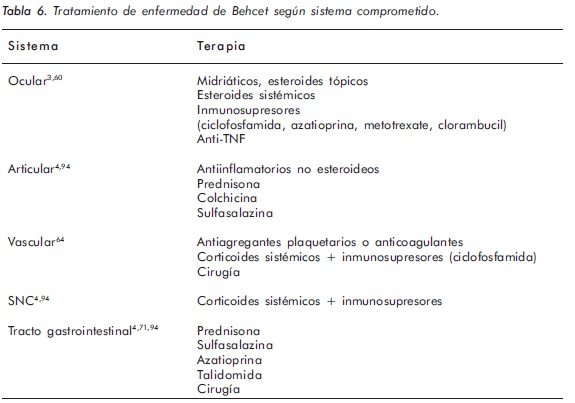
Los avances recientes en el conocimiento de la patogénesis de la EB han llevado a nuevas aproximaciones terapéuticas y a un mejor entendimiento de la acción de los tratamientos establecidos. Por otro lado, la disponibilidad de los nuevos agentes antiinflamatorios e inmunomoduladores, tales como los inhibidores del TNF-α y el interferón α-2a han proporcionado herramientas promisorias en el tratamiento de enfermedad recalcitrante95. Las manifestaciones vasculares y neurológicas aún constituyen desafíos terapéuticos, por lo que se requiere un mayor número de estudios que permitan dilucidar mejores estrategias de tratamiento en enfermedad grave.
Conclusión
La EB es un desorden multisistémico con manifestaciones clínicas variables, cuya etiopatogenia no es muy bien conocida todavía. Se caracteriza por un curso clínico con exacerbaciones y remisiones de duración y frecuencia impredecibles. El tratamiento es usualmente sintomático y paliativo, y requiere manejo multidisciplinario que incluye reumatología, dermatología, oftalmología, neurología y cirugía vascular, entre otras especialidades.
Referencias
1. Al-Mutawa SA, Hegab SM. Behcet's disease. Clin Exp Med 2004; 4: 103-131. [ Links ]
2. Al-Otaibi LM, Porter SR, Poate TW. Behcet's disease: A review. J Dent Res 2005; 84(3): 209-222. [ Links ]
3. Bonfioli A, Orefice F. Behcet's disease. Semin Ophthalmol 2005; 20: 199-206. [ Links ]
4. Kaklamani V, Vaiopoulos G, Kaklamanis P. Behcet's disease. Arthritis Rheum 1998; 27(4): 197-217. [ Links ]
5. Ghate JV, Jorizzo JL. Behçet's disease and complex aphthosis. J Am Acad Dermatol 1999; 40: 1-18. [ Links ]
6. Yurdakul S, Hamuryudan V, Yazici H. Behcet syndrome. Curr Opin Rheumatol 2003; 16: 38-42. [ Links ]
7. Hirohata S, Kikuchi H. Behcet's disease. Arthritis Res Ther 2003; 5: 139-146. [ Links ]
8. Hippocrates. Third book on epidemiology. Case 7. Kaktos (ed). 993; 13: 209. [ Links ]
9. Lisse JR, Oberto-Medina M. 2006. Behcet Disease [en línea] http://www.emedicine.com/med/topic218.htm [consulta 29/07/2006] [ Links ]
10. Tuzun Y, Yurdakul S, Cem MM, Ozyazgan Y, Hamuryudan V, Tuzun B, et al. Epidemiology of Behcet's syndrome in Turkey. Int J Dermatol 1996; 35: 618-620. [ Links ]
11. Jaber L, Milo G, Halpern GJ, Krause I, Weinberger A. Prevalence of Behcet's disease in an Arab community in Israel. Ann Rheum Dis 2002; 61: 365-366. [ Links ]
12. Al Rawi ZS, Neda AH. Prevalence of Behcet's disease among Iraqis. Adv Exp Med Biol 2003; 528: 37-41. [ Links ]
13. Mizuki N, Ota M, Yabuki K, Katsuyama Y, Ando H, Palimeris GD, et al. Localization of the pathogenic gene of Behcet's disease by microsatellite analysis of three different populations. Invest Ophthalmol Vis Sci 2000; 41: 3702-3708. [ Links ]
14. Zouboulis CC, Kotter I, Djawari D, Kirch W, Kohl PK, Ochsendorf FR, et al. Epidemiological features of Adamantiades - Behcet's disease in Germany and in Europe. Yonsei Med J 1997; 38: 411-422. [ Links ]
15. Gonzalez-Gay MA, Garcia-Porrua C, Branas F, Lopez- Lazaro L, Olivieri I. Epidemiologic and Clinical aspects of Behcet's disease in a defined area of Northwestern Spain, 1988-1997. J Rheumatol 2000; 27: 703-707. [ Links ]
16. Londoño A, Hernández D, Palacios C, Castaño H, Montoya C, Donado J, et al. Enfermedad de Behcet: espectro clínico de ocho pacientes en la ciudad de Medellín. Rev Col Reumatol 2003; 10 (3): 199-205. [ Links ]
17. Zouboulis CC, Vaiopoulos G, Marcomichelakis N, Palimeris G, Markidou I, Thouas B, et al. Onset signs, clinical course, prognosis, treatment and aoutcome of adult patients with Adamantiades-Behcet disease in Greece. Clin Exp Rheumatol 2003; 21(4) suppl 30: S19-S26. [ Links ]
18. Jacyk WK. Behçet's disease in South African blacks: report of five cases. J Am Acad Dermatol 1994; 30: 869-873. [ Links ]
19. Gül A. Behcet's disease: An update on the pathogenesis. Clin Exp Rheumatol 2001; 19(suppl. 24): S6-S12. [ Links ]
20. Fietta P. Behcet's disease: Familial clustering and immunogenetics. Clin Exp Rheumatol 2005; 23(4) suppl. 38: S96-S105. [ Links ]
21. Gül A, Inanc M, Ocal L, Aral O, Konice M. Familial aggregation of Behcet's disease in Turkey. Ann Rheum Dis 2000; 59: 622-625. [ Links ]
22. Kóne-Paut I, Geisler I, Wechsler B, Ozen S, Ozdogan H, Rozenbaum M, et al. Familial aggregation in Behcet's disease: high frequency in siblings and parents of pediatric probando. J Pediatr 1999; 135: 89-93. [ Links ]
23. Birdstewart JA. Genetic analysis of families of patients with Behcet's syndrome: data incompatible with autosomal recessive inheritance. Ann Rheum Dis 1986; 45: 265-268. [ Links ]
24. Fresko I, Soy M, Hamuryudan V, Yurdakul S, Yavuz S, Tümer Z, et al. Genetic anticipation in Behcet's syndrome. Ann Rheum Dis 1998; 57: 45-48. [ Links ]
25. Zwirner NW, Dole K, Stastny P. Differential surface expresión of MICA by endotelial cells, fibroblast, keratinocytes, and monocytes. Hum Immunol 1999; 60: 323-330. [ Links ]
26. Ota M, Mizuki N, Katsuyamay, Tamiya G, Shiina T, Oka A, et al. The critical region for Behcet's disease in the human major histocompatibility complex is reduced to 46-kb segment centromeric of HLA - B, by association analysis using refined microsatellite mapping. Am J Hum Genet 1999; 64: 1406-1410. [ Links ]
27. Misuki N, Ota M, Katsuyama Y, Yabuki K, Ando H, Goto K, et al. Association analysis between the MIC-A and HLA - B alleles in Japanese patients with Behcet's disease. Arthritis Rheum 1999; 42: 1961-1966. [ Links ]
28. Wallace GR, Verita DH, Delamaine LJ, Ohno S, Inoko H, Ota M, et al. MIC- A allele profiles and HLA class I associations in Behcet's disease. Immunogenetics 1999; 49: 613-617. [ Links ]
29. Sano K, Yabuki K, Imagawa Y, Shiina T, Mizuki N, Ohno S, et al. The absence of disease-specific polymorphisms within the HLA-B51 gene that is the susceptible locus for Behcet's disease. Tissue Antigens 2001; 58: 77-82. [ Links ]
30. Gül A, Hajeer AH, Worthington J, Barret JH, Ollier WE, Silman AJ. Evidence for linkage of the HLA-B locus in Behcet's disease, obtained using the transmisión disequilibrium test. Arthritis Rheum 2001; 44: 239-241. [ Links ]
31. Hegab S, Al Mutawa S. Immunopathogenesis of Behcet's disease. Clinical Immunol 2000; 96: 174-186. [ Links ]
32. Lee S, Bang D, Cho YH, Lee ES, Sohn S. Polymerase chain reaction reveals herpes simplex virus DNA of patients with Behcet's disease. Arch Dermatol Res 1996; 288: 179-183. [ Links ]
33. Sohn S, Lee ES, Bang D, Lee S (1998). Behcet's disease-like symptoms induced by Herpes simplex virus in ICR mice. Eur J Dermatol 8: 21-23. [ Links ]
34. Baskan EB, Yilmaz E, Saricaoglu H, Alkan G, Ercan I, Mistik R. Detection of parvovirus B19 DNA in the lesional skin of patients with Behcet's disease. Clin Exp Dermatol 2007; 32: 186-190. [ Links ]
35. Cooper C, Pippard EC, Sharp H, Wickham C, Chamberlain MA, Barker DJ. Is Behcet's disease triggered by childhood infection. Ann Rheum Dis 1989; 48: 421-423. [ Links ]
36. Narikawa S, Suzuki Y, Takahashi M, Furukawa A, Sakane T, Mizushima Y. Streptococcus oralis previously identified as uncommon Streptococcus sanguis in Behcet's disease. Arch Oral Biol 1995; 40: 685-690. [ Links ]
37. Direskeneli H, Saruhan-Direskeneli G. The role of heat shock proteins in Behcet's disease. Clin Exp Rheumatol 2003; 21(4) suppl. 30: S44-S48. [ Links ]
38. Emmi L, Brugnolo E Salvati G, Marchione T. Immunopathological aspects of Behcet's disease. Clin Exp Rheumatol 1995; 13: 687-691. [ Links ]
39. Al-Dalaan A, Al-Sedairy S, Al-Balaa S, Al-Janadi M, Elramahi K, Bahabri S, et al. Enhanced interleukin 8 secretion in circulation of patients with Behcet's disease. J Rheumatol 1995; 22: 904-907. [ Links ]
40. Turan B, Gallati H, Erdi H, Gurler A, Michel BA, Villiger PM. Systemic levels of the T cell regulatory cytokines IL- 10 and IL-12 in Behcet's disease: soluble TNFR-75 as a biological marker of disease activity. J Rheumatol 1997; 24: 128-132. [ Links ]
41. Yamashita N, Kaneoka H, Kaneko S, Takeno M, Oneda K, Koizumi H, et al. Role of gamma delta T lymphocytes in the development of Behcet's disease. Clin Exp Immunol 1997; 107: 241-247. [ Links ]
42. Sahin S, Lawrence R, Direskeneli H, Hamuryudan V, Yazici H, Akoglu T. Monocyte activity in Behcet's disease. Br J Rheumatol 1996; 35: 424-429. [ Links ]
43. Haznedaroglu IC, Ozcebe OI, Ozdemir O, Celik I, Dundar SV, Kirazli S. Impaired haemostatic kinetics and endothelial function in Behcet's disease. J Intern Med 1996; 240: 181-187. [ Links ]
44. Gül A, Özbek U, Öztürk C, Inanc M, Konice M, Özcelik T. Coagulation factor V gene mutation increases the risk of venous thrombosis in Behcet's disease. Br J Rheumatol 1996; 35: 1178-1180. [ Links ]
45. Gül A, Aslantas AB, Tekinay T, Konice M, Özcelik T. Procoagulant mutations and venous thrombosis in Behcet's disease. Rheumatology 1999; 38: 1298-1299. [ Links ]
46. Mader R, Ziv M, Adawi M, Mader R, Lavi I. Thrombophilic factors and their relation to thromboembolic and other clinical manifestations in Behcet's disease. J Rheumatol 1999; 26(11): 2404-2408. [ Links ]
47. Alpsoy E, Donmez L, Onder M, Gunasti S, Usta A, Karincaoglu Y, et al. Clinical features and natural course of Behcet's disease in 661 cases: a multicentre study. Br J Dermatol 2007; 157 (5): 901-906. [ Links ]
48. Tursen U, Gürler A, Boyvat A. Evaluation of clinical findings according to sex in 2313 Turkish patients with Behcet's disease. Int J Dermatol 2003; 42: 346-351. [ Links ]
49. Main DM, Chamberlain MA. Clinical differentiation of oral ulceration in Behcet's disease. Br J Rheumatol 1992; 31(11): 767-770. [ Links ]
50. The International Study Group for Behcet's Disease. Evaluation of diagnostic (classification) criteria in Behcet's disease - towards internationally agreed criteria. Br J Rheumatol 1992; 31(5): 299-308. [ Links ]
51. Krause I, Leibovici L, Guedj D, Molad Y, Uziel Y, Weinberger A. Disease patterns of patients with Behcet's disease demonstrated by factor analysis. Clin Exp Rheumatol 1999; 17 (3): 347-350. [ Links ]
52. Magro CM, Crowson AN. Cutaneous manifestations of Behcet's disease. Int J Dermatol 1995; 34: 159-165. [ Links ]
53. King R, Crowson AN, Murray E, Magro CM. Acral purpuric papulonodular lesions as a manifestation of Behcet's disease. Int J Dermatol 1995; 34(3): 190-192. [ Links ]
54. Oguz O, Serdaroglu S, Turzun Y, Erdogan N, Yazici H, Savaskan H. Acute febrile neutrophilic dermatosis (Sweet's syndrome) associated with Behcet's disease. Int J Dermatol 1992; 31(9): 645-646. [ Links ]
55. Takeda U, Kureda K, Shinkai H. Encapsulated necrosis associated with Behcet's disease. J Dermatol 1999; 26(8): 522-526. [ Links ]
56. Inaloz HS, Evereklioglu C, Unal B, Kirtak N, Eralp A, Inaloz SS. The significance of immunohistochemistry in the skin pathergy reaction of patients with Behçet's syndrome. JEADV 2004; 18: 56-61. [ Links ]
57. Dilsen N, Konice M, Aral O, Ocal L, Inanc M, Gul A. Comparative study of the skin pathergy test with blunt and sharp needles in Behcet's disease: confirmed specificity but decreased sensitivity with sharp needles. Ann Rheum Dis 1993; 52: 823-825. [ Links ]
58. Davatchi F, Shahram F, Nadji A, Jamshidi AR, Chams C, Chams H, et al. Influence of pathergy test on the accuracy of different diagnosis criteria for Behçet's disease. Adv Exp Med Biol 2003; 528: 109-112. [ Links ]
59. Tunal-Tutkun I, Onal S, Altan-Yaycioglu R, Huseyin H, Urgancioglu. Uveitis in Behcet disease: an analysis of 880 Patients. Am J Ophthalmol 2004; 138: 373-380. [ Links ]
60. Okada A. Behcet's disease: general concepts and recent advances. Curr Opin Ophthalmol 2006; 17: 551-556. [ Links ]
61. Ben-Ezra D, Cohen E. Treatment and visual prognosis in Behcet's disease. Br J Ophthalmol 1986; 70: 589-592. [ Links ]
62. Sakamoto M, Akasawa K, Nishioka Y, Sanui H, Inomata H, Nose Y. Prognostic factors of vision in patients with Behcet's disease. Opthalmology 1995; 102(2): 317-321. [ Links ]
63. Lie JT. Vascular involvement in Behcet's disease; arterial and venous and vessels of all sizes. J Rheumatol 1992; 19: 341-342. [ Links ]
64. Alpagut U, Ugurlucan M, Dayioglu E. Major arterial involvement and review of Behcet's disease. Ann Vasc Surg 2007; 21(2): 232-239. [ Links ]
65. Hamza M. Large artery involvement in Behcet's disease. J Rheumatol 1987; 14: 554-559. [ Links ]
66. Benekli M, Gullu IH, Haznedaroglu JC. Ischemic heart disease in Behcet's syndrome. Rheumatol Int 1998; 17(6): 251. [ Links ]
67. Haghighi AB, Pourmand R, Nikseresht A. Neuro-Behcet disease: a review. Neurologist 2005; 11(2): 80-89. [ Links ]
68. Akman - Demir G, Serdaroglu P, Tasci B and the Neuro- Behcet study group. Brain 1999; 122: 2171-2181. [ Links ]
69. Diri E, Mat C, Hamuryudan V, Yurdakul S, Hýzlý N, Yazýcý N. Papulopustular skin lesions are seen more frequently in patients with Behcet's syndrome who have arthritis: a controlled and masked study. Ann Rheum Dis 2001; 60: 1074-1076. [ Links ]
70. Yardakul S, Yazici H, Tuzun Y, Pazarli H, Yalcin B, Altac M, et al. The arthritis of Behcet's disease: A prospective study. Ann Rheum Dis 1983; 42: 505-515. [ Links ]
71. Yazici H, Fresko I, Yurdakul S. Behçet's syndrome: disease manifestations, management, and advances in treatment. Nat Clin Pract Rheumatol 2007; 3(3): 148-155. [ Links ]
72. Erkan F, Kiyan E, Tunaci A. Pulmonary complications of Behcet's disease. Clin Chest Med 2002; 23: 493-503. [ Links ]
73. Bayraktar Y, Balkanci F, Bayraktar M, Calguneri M. Budd-Chiari syndrome: a common complication of Behcet's disease. Am J Gastroenterol 1997; 92: 858- 862. [ Links ]
74. Rugby AS, Chamberlain MA, Bhakta B. Classification and assessment of rheumatic diseases: Part 1. Behcet's disease. In: Baillieres Clin Rheumatol 1995; 9: 375-395. [ Links ]
75. Chun SI, Su WP, Lee S. Histopathologic study of cutaneous lesions in Behcet's syndrome. J Dermatol 1990; 17(6): 333-341. [ Links ]
76. Jorizzo JL. Behcet's disease: an update based on the 1985 conference in London. Arch Dermatol 1986; 122: 556-558. [ Links ]
77. Behcet's disease. En: Mckee PH, Calonje E, Granter SR. Pathology of the skin with clinical correlations. 3 Ed. Elsevier Mosby. 2005 p. 685-689. [ Links ]
78. Chen K, Kawahara Y, Miyakawa S, Nishikawa T. Cutaneous vasculitis in Behcet's disease: a clinical and histopathologic study of 20 patients. J Am Acad Dermatol 1997; 36: 689-696. [ Links ]
79. Lawton G, Bhakta BB, Chamberlain MA y Tennant A. The Behçet's Disease Activity Index. Rheumatology 2004; 43(1): 73-78. [ Links ]
80. Bhakta BB, Brennan P, James TE, Chamberlain MA, Noble BA, Silman AJ. Behcet's disease: evaluation of a new instrument to measure clinical activity. Rheumatology 1999; 38: 728-733. [ Links ]
81. Yacizi H, Tüzün Y, Pazarli H, Yurdakul S, Ozyazgan H, Ozdogan S, et al. Influence of age of onset and patient's sex on the prevalence and severity of manifestations of Behcet's syndrome. Ann Rheum Dis 1984; 43: 783-789. [ Links ]
82. Krause I, Uziel Y, Guedj D, Mukamel M, Harel L, Molad Y, et al. Childhood Behcet's disease: clinical features and comparison with adult-onset disease. Reumatology 1999; 38: 457-462. [ Links ]
83. Bardak Y. Effects of age and sex on Behçet's disease. J Rheumatol 1999; 26: 1008-1009. [ Links ]
84. Demiroglu H, Dundar S. Effects of age, sex, and initial presentation on the clinical course of Behçet's syndrome. South Med J 1997; 90: 567. [ Links ]
85. Bang DS, Oh SH, Lee KH, Lee ES, Lee SN. Influence of sex on patients with Behçet's disease in Korea. J Korean Med Sci 2003; 18: 231-235. [ Links ]
86. Kural-Seyahi E, Fresko I, Seyahi N, Ozyazgan Y, Mat C, Hamuryudan V, et al. The long-term mortality and morbidity of Behçet syndrome: a 2-decade outcome survey of 387 patients followed at a dedicated center. Medicine (Baltimore) 2003; 82: 60-76. [ Links ]
87. Lin P, Liang G. Behcet disease: Recommendation for clinical management of mucocutaneous lesions. J Clin Rheumatol 2006; 12: 282-286. [ Links ]
88. Ravitskiy L, Green JJ, Goldenberg G, Jorizzo JL. Behcet's disease. In: Lebwohl MG, Heymann WR, Berth-Jones J, Coulson L. Treatment of skin disease comprehensive therapeutic strategies 2ed. Elsevier Mosby. 2006, p. 82-85. [ Links ]
89. Alpsoy E, Er H, Durusoy C, Yilmaz E. The use of sucralfate suspension in the treatment of oral and genital ulceration of Behcet's disease. Arch Dermatol. 1999; 135: 529-532. [ Links ]
90. Jorizzo JL, Hudson RD, Schmalstieg FC, Daniels JC, Apisarnthanarax P, Henry JC, et al. Behçet's syndrome: immune regulation, circulating immune complexes, neutrophil migration, and colchicine therapy. J Am Acad Dermatol 1984; 10: 205-214. [ Links ]
91. Sharquie K, Najim R, Abu-Raghif A. Dapsone in Behcet's disease: a double-blind, placebo-controlled, cross-over study. J Dermatol. 2002; 29: 267-279. [ Links ]
92. Chang ME, Liang GC. Pentoxifylline use for Behcet's disease. Arthritis Rheum 2000; 43: S363. [ Links ]
93. Chang EM, Liang GC. Pentoxifylline use for Behcet's disease: a follow- up cohort study. Arthritis Rheum 2001; 44: S120. [ Links ]
94. Goker B, Goker H. Current therapy for Behcet's disease. Am J Ther 2002; 9(5): 465-470. [ Links ]
95. Pipitone N, Olivieri I, Cantini F, Triolo G, Salvarani C. New approaches in the treatment of Adamantiades- Behcet's disease. Curr Opin Rheumatol 2006; 18: 3-9. [ Links ]

