










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Colombiana de Reumatología
versión impresa ISSN 0121-8123
Rev.Colomb.Reumatol. v.16 n.1 Bogotá ene./mar. 2009
PRESENTACIÓN DE CASO
Churg-Strauss syndrome: a case with unusual manifestations
Mauricio Restrepo1, Luis Alonso González2, Gloria Vásquez2, David Londoño3,
Luis Alfonso Correa4, Luis Alberto Ramírez2
1 Médico internista, residente de reumatología. Universidad de Antioquia.
2 Profesores Sección de Reumatología. Hospital Universitario San Vicente de Paúl, Universidad de Antioquia. Medellín, Colombia.
3 Residente de dermatología. Universidad de Antioquia.
4 Dermatopatólogo. Profesor Sección de Dermatología. Hospital Universitario San Vicente de Paúl. Universidad de Antioquia.
Recibido: Octubre 18/2008 Aceptado: Enero 18/2009
Resumen
El síndrome de Churg-Strauss, una vasculitis sistémica necrotizante que compromete vasos de pequeño y, rara vez, de mediano calibre, es junto con la granulomatosis de Wegener y la poliangeítis microscópica una de las vasculitis asociadas con anticuerpos contra el citoplasma de los neutrófilos. Se caracteriza por asma, hipereosinofilia y granulomas extravasculares eosinofílicos. Reportamos el caso de una mujer de 36 años con historia de asma de comienzo tardío y rinitis alérgica quien presentó compromiso del sistema nervioso central, neuropatía periférica, vasculitis leucocitoclástica y marcada eosinofilia. De manera interesante, esta paciente presentó manifestaciones inusuales del síndrome de Churg-Strauss tales como microaneurismas mesentéricos y claudicación mandibular. Además, presentamos una breve revisión de la literatura sobre síndrome de Churg-Strauss.
Palabras clave: vasculitis, síndrome de Churg-Strauss, ANCA, microaneurismas mesentéricos.
Summary
Churg-Strauss syndrome, a necrotizing systemic vasculitis which involves the small and (more rarely) the medium-sized vessels, is a primary vasculitis strongly associated with antineutrophil cytoplasm antibodies (ANCA). It is characterized by the presence of asthma, eosinophilia and extravascular eosinophilic granulomas. Herein, we report a 36-year-old woman with a history of late onset asthma and allergic rhinitis who developed central nervous system involvement, peripheral neuropathy, leukocytoclastic vasculitis and eosinophilia. Interestingly, unusual clinical manifestations of Churg-Strauss syndrome such as mesenteric microaneurysms and jaw claudication were present in this particular patient. A brief review of the literature of Churg-Strauss syndrome is presented.
Key words: vasculitis; Churg-Strauss syndrome, ANCA, mesenteric microaneurysms.
Introducción
Las vasculitis sistémicas primarias son enfermedades poco frecuentes en la población general. Sus manifestaciones clínicas variadas con síntomas insidiosos pueden generar retardo en el diagnóstico con graves secuelas para el paciente. El reconocimiento de las características típicas y atípicas de los distintos casos evaluados permite una aproximación más acertada y oportuna al enfoque diagnóstico y terapéutico. Presentamos el caso de una mujer con síndrome de Churg-Strauss (SCS) y hacemos una revisión de la literatura de esta entidad resaltando las manifestaciones poco usuales.
Presentación de caso
Mujer de 36 años, natural de Medellín, quien fue hospitalizada en julio de 2008 por dolor abdominal y compromiso neurológico. Con antecedente de asma y rinitis alérgica, ambas diagnosticadas a los 34 años, y tratada con broncodilatadores inhalados. Sus síntomas respiratorios duraron aproximadamente dos años y mejoraron por completo seis meses antes de la consulta actual. En enero de 2008 presentó hipoestesia en miembro inferior izquierdo y un mes después hipoestesia y ardor en mano izquierda, así como hipoestesia, frialdad, dolor "urente" y hemiparesia en el hemicuerpo derecho, con gran limitación para la marcha. En abril de 2008, presentó brote purpúrico en miembros inferiores y dolor abdominal difuso, que inicia en epigastrio, irradia a la espalda, y empeora con la alimentación. Además de lo anterior, pérdida de 11 kg de peso en seis meses, disminución de agudeza visual por ojo derecho, claudicación de mandíbula y artralgias en rodillas.
Al examen físico, alerta y sin dificultad respiratoria. FC 108', PA 120/90, FR 16, afebril (36.5°C), 35 kg de peso. Sin hallazgos anormales sobre las arterias temporales. Fundoscopia bilateral normal. Pequeñas adenopatías cervicales indoloras. Examen cardiopulmonar normal. Abdomen blando, sin masas ni soplos. Pulsos periféricos y centrales fuertes y simétricos. Hipoestesia en hemicuerpo derecho y en pierna y región palmar izquierda. Hiperreflexia y hemiparesia derecha (3/5); marcha parética y antálgica. Púrpura palpable en 2/3 distales de piernas y pies (Figura 1).
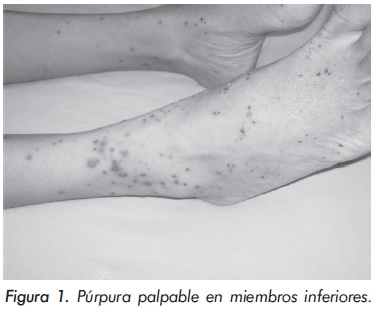
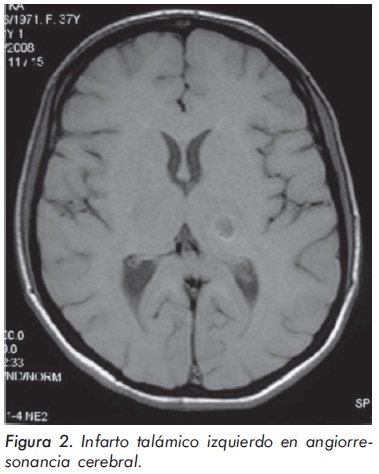
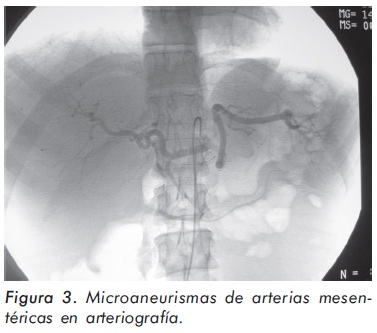
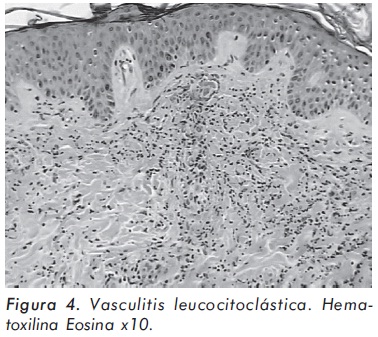
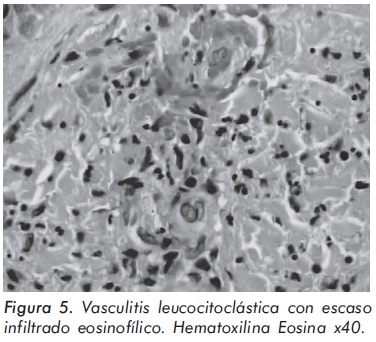
El cuadro hemático inicial mostró leucocitosis (31.500/mm3), eosinofilia (14.300/mm3), trombocitosis (935.000/mm3) y hemoglobina de 12.1 g/ dl; elevación de reactantes de fase aguda (velocidad de sedimentación globular de 55 mm/h, proteína C reactiva de 6,3 mg/dl) y elevación de transaminasas (AST 77 U/L y ALT 64 U/L). Los anticuerpos contra el citoplasma de los neutrófilos Figura 1. Púrpura palpable en miembros inferiores. (ANCAS) y ANAS fueron negativos, al igual que los marcadores virales para VIH, hepatitis B y hepatitis C. La función renal y el uroanálisis fueron normales al igual que los rayos X de tórax. La electromiografía y neuroconducción de cuatro extremidades demostró la presencia de neuropatía sensitivo-motora axonal de predominio en miembros inferiores. Los hallazgos en la ecocardiografía transtorácica fueron una presión sistólica de la arteria pulmonar de 34 mm Hg, leves insuficiencias aórtica, mitral y tricuspídea. En la angiorresonancia cerebral se evidenció un infarto talámico derecho antiguo (Figura 2). Los hallazgos de la arteriografía de vasos abdominales con cateterización selectiva de arterias mesentéricas, renales y tronco celiaco fueron: estenosis multisegmentarias en la arteria mesentérica superior con microaneurismas distales en vasos yeyunales y microaneurismas en capilares distales de la arteria mesentérica inferior (Figura 3). La biopsia de piel demostró vasculitis leucocitoclástica con escaso infiltrado eosinofílico (Figuras 4 y 5). La histología de la biopsia de nervio sural fue normal.
Con base en la presencia de asma de aparición tardía con posterior mejoría, púrpura palpable (vasculitis leucocitoclástica), marcada eosinofilia así como el compromiso neurológico tanto central como periférico y de vasos mesentéricos con formación de microaneurismas se hizo un diagnóstico de SCS. Se inició inmunosupresión con pulsos intravenosos de ciclofosfamida (1 gramo mensual) y metilprednisolona intravenosa (3 dosis de 500 mg cada una) con buena respuesta luego de seis meses de tratamiento, caracterizada por desaparición de sus síntomas abdominales, normalización de las cifras de eosinófilos y recuperación completa de su hemiparesia y una marcha normal.
Síndrome de Churg-Strauss
Es una enfermedad poco frecuente, asociada con ANCA, que afecta vasos sanguíneos de pequeño calibre de pulmones, nervios periféricos y piel, y menos frecuentemente del corazón y tracto gastrointestinal. Clínicamente se manifiesta con síndromes alérgicos como asma, rinitis y sinusitis, y comparte ciertas características clínicas y patológicas con granulomatosis de Wegener (GW) y poliarteritis nodosa (PAN)1,2.
La descripción original de esta entidad, hecha en 1951 por los patólogos Jacob Churg y Lotte Strauss, consistió en el reporte de los hallazgos de autopsia de 13 pacientes que presentaron un síndrome clínico caracterizado por asma, hipereosinofilia y vasculitis sistémica. Las tres principales características histológicas encontradas durante el examen patológico de estos casos fueron granulomas extravasculares, eosinofilia tisular y vasculitis necrosante3.
Sin embargo, varias series de casos posteriormente demostraron que no todos los pacientes tienen las tres principales características histológicas, por lo cual para facilitar el diagnóstico clínico temprano, diferenciarla de otras vasculitis sistémicas e iniciar el manejo oportuno y apropiado se han propuesto varias definiciones y esquemas de clasificación para la enfermedad2 (Tabla 1).

Desde las primeras descripciones ha existido controversia sobre la verdadera identidad del SCS y su distinción de la PAN; sin embargo, en la actualidad se considera una enfermedad bien caracterizada con base en sus particularidades clínicas e histológicas4.
Epidemiología
La PAN clásica fue descrita por primera vez en 1866 por Kussmaul y Meyer como una condición caracterizada por inflamación y necrosis de vasos de mediano calibre que lleva a la formación de aneurismas e infarto de órganos. A pesar de que durante diferentes momentos del siglo XX se caracterizaron y se separaron de la PAN entidades distintas como la GW, el SCS y la poliangeítis microscópica (PAM), todavía en 1980 se usaba el término genérico de PAN para describir cualquier forma de vasculitis sistémica primaria de pequeño y mediano vaso, lo cual hace difícil obtener una información epidemiológica precisa5.
La mayor parte de la información epidemiológica proviene de países europeos donde se han encontrado incidencias anuales de 10 a 20, y prevalencias entre 90 y 257 casos por millón para el grupo de vasculitis asociadas con ANCA, siendo la menos frecuente de las tres (GW , PAM, SCS), el SCS5. Se ha reportado una incidencia anual de 2,4 a 4 casos por millón y un ligero predominio en la población masculina2. SCS usualmente se presenta entre los 14 y 75 años, con un promedio de edad de 50 años. Existen pocos casos reportados en niños6, 7.
Patogénesis
Las vasculitis difieren no solo en su presentación clínica y patológica, sino también en su presunta patogénesis. Las vasculitis de vasos grandes son causadas supuestamente por reacciones inmunes mediadas por células contra autoantígenos pobremente definidos. En algunas vasculitis de pequeños vasos (púrpura de Henoch Schönlein, crioglobulinemia, angeítis leucocitoclástica) se han encontrado depósitos de complejos inmunes. Por el contrario, las lesiones vasculíticas causadas por GW, PAM y SCS, frecuentemente carecen de depósitos inmunes por lo cual son consideradas como vasculitis necrosantes pauciinmunes, y se caracterizan por la presencia de ANCA8. Una cuarta forma de vasculitis asociada con ANCA es la glomerulonefritis crescéntica necrosante pauciinmune limitada al riñón, que se considera a veces una forma limitada de PAM9.
Los ANCA fueron descritos inicialmente a principios de los años ochenta en asociación con vasculitis y glomerulonefritis pauciinmune. Posteriormente se identificó a los antígenos proteinasa 3 y mieloperoxidasa como los responsables de los patrones citoplasmático y perinuclear respectivamente observados en inmunofluorescencia sobre neutrófilos fijados con etanol. A pesar de existir controversia sobre si los ANCA tienen un papel fisiopatológico o son una manifestación más del fenómeno inflamatorio, múltiples evidencias de origen animal y observaciones clínicas en humanos apoyan la hipótesis de que estos autoanticuerpos son facilitadores y perpetuadores de una respuesta inmune anormal10.
En el SCS solo el 40% de los pacientes es ANCA positivo con especificidad para mieloperoxidasa. De manera interesante aquellos pacientes con SCS ANCA positivo presentan vasculitis de pequeño vaso que se manifiesta por mononeuritis múltiple, púrpura y glomerulonefritis, mientras que aquellos con SCS ANCA negativo predominantemente muestran infiltración tisular por eosinófilos, características estas que no comparte nuestra paciente. Esta observación sugiere que el SCS puede agrupar al menos dos entidades clínicas distintas8, 11-13.
Manifestaciones clínicas
Se han descrito tres fases de evolución de la enfermedad parcialmente superpuestas. Una primera fase prodrómica caracterizada por manifestaciones atópicas como asma, rinitis alérgica, sinusitis y pólipos nasales. El asma se caracteriza por ser de aparición tardía (en el adulto), y puede llegar a ser refractaria al manejo convencional. Puede preceder la aparición de la vasculitis entre tres y ocho años; sin embargo, se ha descrito un periodo prodrómico hasta de 30 años. Tal como fue reportado en el artículo original por Churg y Strauss, y como sucedió en nuestra paciente, hasta la mitad de los pacientes pueden presentar notable mejoría o incluso dramática resolución de su cuadro de asma antes del inicio de la fase vasculítica2, 3. Existen algunos reportes en pacientes no asmáticos en quienes las manifestaciones vasculíticas precedieron la aparición de asma14.
La segunda fase se caracteriza por eosinofilia periférica, y pueden ocurrir infiltrados eosinofílicos en pulmones y tracto gastrointestinal. La eosinofilia usualmente es mayor del 10% del conteo leucocitario o mayor de 1500/mm3. En esta fase se presentan síntomas constitucionales como fiebre y pérdida de peso. Durante la tercera fase de la enfermedad ocurren manifestaciones vasculíticas. Además de los síntomas constitucionales también hay manifestaciones osteomusculares como mialgias, artralgias y artritis. Los reactantes de fase aguda se encuentran elevados por lo general. Se han descrito casos de compromiso limitado a un solo órgano2, 15.
El compromiso neurológico es frecuente, siendo la neuropatía periférica mucho más prevalente que en las otras vasculitis de pequeños vasos asociadas con ANCA. La neuropatía periférica en el SCS ocurre en un 50% a 75% de los casos. Típicamente ocurre mononeuritis múltiple ocasionada por la lesión vasculítica de la vasa nervorum. Ocasionalmente la lesión neuropática periférica puede tener alguna mejoría con la terapia pero en general el daño suele ser permanente y causando importante morbilidad y discapacidad funcional. Aunque el compromiso del sistema nervioso central es menos frecuente, es la segunda causa de muerte en SCS y se presenta con infarto o hemorragia cerebral2, 16.
El compromiso gastrointestinal puede manifestarse con un abdomen agudo o dolor anginoso postprandial, colecistitis, hemorragia y perforación intestinal2. La presencia de microaneurismas en circulación esplácnica con cuadros de isquemia mesentérica ha sido descrita; en una serie de 96 pacientes con SCS se encontraron microaneurismas en 9 de 26 pacientes a quienes se les realizó angiografía celiaco-mesentérica y/ o renal17. Este inusual hallazgo fue demostrado en nuestra paciente.
Las manifestaciones cutáneas se presentan en aproximadamente el 50% de los pacientes, siendo las más frecuentes, la púrpura palpable y las petequias en las extremidades inferiores, y los nódulos y pápulas en superficies extensoras de las articulaciones, más comúnmente en codos. Otras lesiones descritas son maculopápulas eritematosas tipo eritema multiforme, úlceras, livedo reticularis y edema facial. La presencia en biopsia de piel de granuloma necrosante extravascular (granuloma Churg- Strauss) se correlaciona clínicamente con lesiones tipo pápulas y nódulos, mientras que lesiones tipo petequias y púrpura se correlacionan con vasculitis leucocitoclástica en la biopsia18, 19.
La enfermedad cardiaca es la principal causa de muerte y es responsable del 50% de los fallecimientos. Puede ocurrir falla cardiaca por miocarditis y asociarse con pericarditis e hipertensión. Con menor frecuencia se ha reportado isquemia coronaria, cardiomiopatía restrictiva y arritmias2,16,20. En pulmón puede haber infiltrados pulmonares, derrame pleural, nódulos que rara vez forman cavernas, y hemorragia pulmonar secundaria a capilaritis21. Las alteraciones renales son inusuales y suelen responder rápidamente al manejo2, 22.
Una manifestación clínica de nuestra paciente y rara vez reportada en la literatura es la claudicación de mandíbula muy posiblemente en relación con arteritis temporal. Este hallazgo fue descrito en uno de los 96 pacientes de la serie de Guillevin17. La presencia de vasculitis de la arteria temporal en personas menores de 50 años ha sido reportada en la literatura en menos de 40 casos y cinco de ellos correspondieron a SCS23. Sin embargo, la confirmación histológica de compromiso de la arteria temporal en esta paciente no fue realizada.
Diagnóstico
El diagnóstico de SCS se basa en datos clínicos. En presencia de asma, rinitis o sinusitis, asociada con eosinofilia periférica y síntomas de vasculitis debe sospecharse fuertemente; sin embargo, es necesaria la histología para confirmar el diagnóstico2.
Los hallazgos histológicos más comunes en el SCS son la vasculitis leucocitoclástica, la cual puede estar acompañada o no por un intenso infiltrado de eosinófilos y el granuloma extravascular necrotizante, antes denominado granuloma de Churg-Strauss. La biopsia de las lesiones cutáneas puede mostrar, además de los hallazgos anteriores, un patrón histológico indistinguible del observado en la PAN cutánea. Ninguno de los hallazgos histológicos anteriores es exclusivo del SCS; la vasculitis leucocitoclástica se encuentra en las vasculitis alérgicas de pequeños vasos y en la GW, mientras que el granuloma extravascular necrotizante se puede encontrar en la GW, el lupus eritematoso sistémico, la artritis reumatoide y diversos procesos linfoproliferativos. Los hallazgos histológicos pueden documentarse en la biopsia de pulmón, tracto gastrointestinal, corazón, piel, y rara vez riñón. De estas, la biopsia de piel es la que presenta una mayor facilidad de realización. La biopsia de músculo y de nervio, especialmente en presencia de neuropatía, también puede aportar información valiosa16,18,19.
La evaluación clínica inicial debe tratar de establecer el diagnóstico con precisión, evaluar el grado de compromiso sistémico, el daño de órganos y el nivel de actividad para definir la terapia óptima y posteriormente evaluar la respuesta a esta1.
Los ANCA se encuentran presentes en menos de la mitad de los pacientes clasificados como SCS, así que su ausencia no descarta la enfermedad. Las fluctuaciones en los títulos de estos autoanticuerpos no se correlacionan estrictamente con la actividad clínica de este grupo de vasculitis; sin embargo, se recomienda prolongar las terapias en caso de persistir positivos, y vigilar estrechamente a aquellos pacientes que vuelven a aumentar sus títulos8, 24.
El diagnóstico diferencial incluye al síndrome hipereosinofílico, la neumonía eosinofílica crónica y otros tipos de vasculitis sistémicas. Otros desórdenes que comparten algunas características con el SCS son granuloma eosinofílico, sarcoidosis, vasculitis eosinofílica cutánea, procesos infecciosos granulomatosos y reacciones alérgicas a medicamentos2,16.
Tratamiento
El manejo se basa en recomendaciones de expertos. La piedra angular del manejo son los glucocorticoides, los cuales inducen y mantienen una respuesta adecuada en la mayoría de los pacientes; sin embargo, algunos requieren el uso de citotóxicos con base en la presencia de ciertas características clínicas y factores de mal pronóstico2, 19.
Los objetivos de tratamiento son inducción de la remisión y luego mantenimiento de la remisión. En general se inicia con esteroides a dosis de 1 mg/kg/día en equivalentes de prednisolona por uno a dos meses o hasta controlar la actividad vasculítica cuando se inicia un descenso gradual. En situaciones graves o amenazantes de órganos vitales o de la vida del paciente se usan bolos de glucocorticoides intravenosos por uno a tres días. En caso de no controlar la vasculitis y de necesitar dosis inaceptablemente altas para el control de la actividad de la enfermedad, o en presencia de factores de mal pronóstico identificados por el grupo de Guillevin debe utilizarse ciclofosfamida2. Estos factores son: compromiso renal dado por proteinuria mayor de 1 gramo o creatinina mayor de 1,6 mg/dl, compromiso cardiaco, gastrointestinal o del sistema nervioso central17,19. Se han utilizado otros medicamentos como azatioprina o metotrexate principalmente durante la fase de mantenimiento o con el objetivo de ahorrar esteroides2. Existen reportes de casos refractarios a esteroides y ciclofosfamida tratados en forma exitosa con interferón alfa o con rituximab25, 26.
Conclusión
Los hallazgos clínicos de esta paciente donde sobresalieron el asma de aparición tardía, la marcada hipereosinofilia, las lesiones en piel, el compromiso de tracto gastrointestinal, sistema nervioso central y nervio periférico, y la confirmación histológica de vasculitis permitió hacer un diagnóstico de síndrome de Churg-Strauss. El SCS es una vasculitis primaria sistémica infrecuente, con características clínicas y de laboratorio usualmente fáciles de identificar, y que en general responde rápidamente al manejo con glucocorticoides. No obstante, por el carácter de compromiso de múltiples órganos, la variedad de manifestaciones clínicas y la posibilidad de compromiso grave de órganos vitales o de la vida del paciente, con formas a veces severas y refractarias al manejo convencional, es necesario efectuar el tratamiento en centros de referencia especializados en estas patologías.
Referencias
1. Keogh KA, Specks U. Churg-Strauss Syndrome. Semin Respir Crit Care Med 2006; 27: 148-157. [ Links ]
2. Abril A, Calamia KT, Cohen MD. The Churg Strauss Syndrome (Allergic Granulomatous Angiitis): Review and Update. Semin Arthritis Rheum 2003; 33: 106- 114. [ Links ]
3. Churg J, Strauss L. Allergic granulomatosis, allergic angiiis, and periarteritis nodosa. Am J Pathol 1951; 27: 277-301. [ Links ]
4. Conron M, Beynon HLC. Churg-Strauss syndrome. Thorax 2000; 55: 870-877. [ Links ]
5. Watts RA, Lane S, Scott DGI. What is known about the epidemiology of the vasculitides? Best Pract & Res Clin Rheum 2005; 19: 191-207. [ Links ]
6. Boyer D, Vargas SO, Slattery D, Rivera-Sanchez YM, Colin AA. Churg-Strauss Syndrome in Children: A Clinical and Pathologic Review. Pediatrics 2006; 118: 914-920. [ Links ]
7. Zwerina J, Eger G, Englbrecht M, Manger B, Schett G. Churg-Strauss Syndrome in Childhood: A Systematic Literature Review and Clinical Comparison with Adult Patients. Semin Arthritis Rheum 2008. Artículo en prensa. doi:10.1016/j.semarthrit. 2008. 05.004 [ Links ]
8. Kallenberg CGM, Heeringa P, Stegeman CA. Mechanisms of Disease: pathogenesis and treatment of ANCA-associated vasculitides. Nat Clin Pract Rheum 2006; 12: 661-670. [ Links ]
9. Belmont HM. Treatment of ANCA-associated Systemic Vasculitis. Bull NYU Hosp Jt Dis 2006; 64: 60-66. [ Links ]
10. Csernok E. Anti-neutrophil cytoplasmic antibodies and pathogenesis of small vessel vasculitides. Autoimmunity Reviews 2003; 2: 158-164. [ Links ]
11. Sinico RA, Di Toma L, Maggiore U, Bottero P, Radice A, Tosoni C, et al. Prevalence and clinical significance of antineutrophil cytoplasmic antibodies in Churg- Strauss syndrome. Arthritis Rheum 2005; 52: 2926- 2935. [ Links ]
12. Sablé-Fourtassou R, Cohen P, Mahr A, Pagnoux C, Mouthon L, Jayne D, et al. Antineutrophil cytoplasmic antibodies and the Churg-Strauss syndrome. Ann Intern Med 2005; 143: 632-638. [ Links ]
13. Kallenberg CGM. Churg-Strauss syndrome: just one disease entity? Arthritis Rheum 52: 2589-2593. [ Links ]
14. Kawakami T, Soma Y, Hosaka E, Mizoguchi M. Churg- Strauss Syndrome with Cutaneous and Neurological Manifestations Preceding Asthma. Acta Derm Venereol 2006; 86: 67-68. [ Links ]
15. Churg A. Recent Advances in the Diagnosis of Churg- Strauss Syndrome. Mod Pathol 2001; 14: 1284-1293. [ Links ]
16. Noth I, Strek ME, Leff AR. Churg-Strauss syndrome. Lancet 2003; 361: 587-594. [ Links ]
17. Guillevin L, Cohen P, Gayraud M, Lhote F, Jarrousse B, Casassus P. Churg-Strauss Syndrome: Clinical Study and Long-Term Follow-Up of 96 Patients. Medicine 1999; 78: 26-37. [ Links ]
18. Davis MDP, Daoud MS, McEvoy MT, Su WPD. Cutaneous manifestations of Churg-Strauss syndrome: A clinicopathologic correlation. J Am Acad Dermatol 1997; 37: 199-203. [ Links ]
19. Pagnoux C, Guilpain P, Guillevin L. Churg-Strauss syndrome. Curr Op Rheumatol 2007; 19: 25-32. [ Links ]
20. Pelà G, Tirabassi G, Pattoneri P, Pavone L, Garini G, Bruschi G. Cardiac Involvement in the Churg-Strauss Syndrome. Am J Cardiol 2006; 97: 1519-1524. [ Links ]
21. Lai RS, Lin SL, Lai NS, Lee PC. Churg-Strauss Syndrome Presenting with Pulmonary Capillaritis and Diffuse Alveolar Hemorrhage. Scand J Rheumatol 1998; 27: 230-232. [ Links ]
22. Murray NB, Barber FA, Murray KM. Asthma, Eosinophilia and Palpable Purpura. Chest 1989; 96: 416-418. [ Links ]
23. Nesher G, Oren S, Lijovetzky G, Nesher R. Vasculitis of the Temporal Arteries in the Young. Sem Arthritis Rheum 2008. Artículo en prensa. doi:10.1016/ j.semarthrit.2008.03.001 [ Links ]
24. Seo P, Stone JH. The Antineutrophil Cytoplasmic Antibody-Associated Vasculitides. Am J Med. 2004; 117: 39-50. [ Links ]
25. Tatsis E, Schnabel A, Gross WL. Interferon-alpha treatment of four patients with the Churg-Strauss syndrome. Ann Intern Med 1998; 129: 370-374. [ Links ]
26. Koukoulaki M, Smith KGC, Jayne DRW. Rituximab in Churg-Strauss síndrome. Ann Rheum Dis 2006; 65: 557-559. [ Links ]

