










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Reumatología
Print version ISSN 0121-8123
Rev.Colomb.Reumatol. vol.16 no.2 Bogotá Apr./June 2009
ARTÍCULO DE REVISIÓN
Urticarial Vasculitis
Ana María Rivas González1, Carlos Jaime Velásquez Franco2, Luis Fernando Pinto Peñaranda2, Javier Darío Márquez2
1 RII Dermatología, Universidad Pontificia Bolivariana. Medellín, Colombia.
2 Docente, Profesor de Reumatología, Universidad Pontificia Bolivariana. Internista Reumatólogo HPTU. Hospital Pablo Tobón Uribe, Medellín, Colombia. Correo electrónico: anirivas@hotmail.com.
Recibido: Abril 15 de 2009 Aceptado: Junio 16 de 2009
Resumen
La urticaria vasculítica (UV) es una entidad clinicopatológica caracterizada por episodios recurrentes de urticaria y vasculitis leucocitoclástica en la histopatología. Una minoría de pacientes con urticaria crónica tiene urticaria vasculítica (aproximadamente un 5%). Aunque la definición de vasculitis ha cambiado de un estudio a otro, en este artículo nos adherimos a los autores que consideran que la leucocitoclasia y los depósitos de fibrina son indispensables para definir la entidad. Clásicamente la UV se manifiesta con habones eritematosos recurrentes por más de cuatro a seis semanas, que duran más de 24 horas y desaparecen dejando hiperpigmentación residual. Como las características de la urticaria vasculítica pueden sobreponerse con las de la urticaria común, su diagnóstico siempre debe apoyarse en un estudio histopatológico. Según los niveles de complemento esta entidad puede subdividirse en urticaria vasculítica normocomplementémica e hipocomplementémica. Una minoría de pacientes con urticaria vasculítica hipocomplementémica cumple con criterios diagnósticos de síndrome de urticaria vasculítica hipocomplementémica. Aquellos pacientes con hipocomplementemia tienen mayor riesgo de desarrollar compromiso multiorgánico y frecuentemente desarrollan lupus eritematoso sistémico (LES) durante el seguimiento, especialmente los que cursan con anticuerpos anti-C1q.
Palabras clave: urticaria, vasculitis, lupus eritematoso sistémico.
Summary
Urticarial vasculitis is a clinic-pathologic entity typified by recurrent episodes of urticaria that have the histopathologic features of leukocytoclastic vasculitis. Only a minority of patients with chronic urticarial lesions have urticarial vasculitis (approximately 5%). Even though the definition of vasculitis has varied, in this article, we adhere to the concept that leukocytoclasis and fibrinoid deposits are the most important features to define this entity. Classically, urticarial vasculitis manifest with recurrent erythematosus wheals that last for more than 4-6 weeks, the individual lesions persist more than 24 hours and leave residual hyperpigmentation. Because clinical characteristics of urticarial vasculitis may overlap with those of common urticaria, confirmation of the diagnosis requires a lesional skin biopsy. Urticarial vasculitis can be classified as normocomplementemic or hypocomplementemic depending on seric complement levels. Only a minority of patients with hypocomplementemic urticarial vasculitis fulfill diagnostic criteria for hypocomplementemic urticarial vasculitis syndrome. Hypocomplementemic patients have the propensity to have more severe multi-organ involvement and frequently develop systemic lupus erythematous when they are followed in time, in special when they have anti-C1q antibodies.
Key words: urticaria, vasculitis, lupus erythematosus systemic.
Introducción
La UV es una entidad clinicopatológica caracterizada por episodios recurrentes de urticaria que se acompañan de vasculitis leucocitoclástica en la histopatología1-3. Aunque en algunos casos corresponde a un fenómeno cutáneo local que no se asocia a una enfermedad subyacente, en otros pacientes se presenta como una manifestación de una enfermedad sistémica2,4.
Según los niveles de complemento esta entidad puede subdividirse en urticaria vasculítica normocomplementémica (UVN) e hipocomplementémica (UVH) 5,6. Solo una minoría de pacientes con UVH cumple con criterios diagnósticos de síndrome de urticaria vasculítica hipocomplementémica (SUVH), que se caracteriza por lesiones recurrentes de UV asociadas a hipocomplementemia, compromiso multiorgánico y anticuerpos contra la región colágena del C1q4.
Por la gran variedad de manifestaciones cutáneas, sistémicas y serológicas, esta patología ha recibido diferentes denominaciones a través del tiempo, tales como hipocomplementemia con vasculitis cutánea y artritis1, urticaria y artralgias con angitis necrotizante7, síndrome de urticaria vasculítica hipocomplementémica8, y síndrome inusual Lupus like9.
Epidemiología
La UV es una entidad relativamente rara. Su incidencia varía dependiendo de los criterios histológicos utilizados para definir la condición10. En casos en los cuales la simple presencia de neutrófilos en los vasos sanguíneos es suficiente para diagnosticar UV, la prevalencia puede ser tan alta como el 20% de los casos de urticaria crónica. Sin embargo, cuando se utilizan criterios más estrictos como la leucocitoclasia y la presencia de depósitos fibrinoides, la incidencia varía entre un 2% y un 20%3,11. Muchos autores concuerdan en que la incidencia aproximada de UV en pacientes con urticaria crónica es aproximadamente un 5%3,10,12.
La UV es más común en el sexo femenino, en el cual se presentan en el 57 al 80% de los casos5-6,13-15. Se presenta principalmente en la cuarta década de la vida5,13,15 y es una entidad poco frecuente en la población pediátrica16-18.
Histopatología
La UV es una manifestación de la inflamación de los capilares y las vénulas post-capilares de la piel2-4. Como en muchos casos las lesiones clínicas de UV son indistinguibles de la urticaria alérgica; la biopsia de piel es el estándar de oro para hacer el diagnóstico de esta entidad2,10. Sin embargo, vale la pena resaltar que no hay un consenso claro sobre los criterios histopatológicos para definir UV y éstos han variado de un estudio a otro10.
La gran mayoría de los autores aceptan que la UV frecuentemente se manifiesta como una vasculitis leucocitoclástica (VL) que se caracteriza por presentar degranulación y fragmentación de neutrófilos que da lugar a la formación de polvo nuclear, edema endotelial, depósitos fibrinoides que pueden llegar a ocluir los vasos sanguíneos, extravasación de eritrocitos, e infiltrado perivascular usualmente rico en neutrófilos. En algunos casos se pueden presentar infiltrados mixtos con linfocitos y eosinófilos4,5,10,11,19.
Las lesiones menores de 24 horas son usualmente ricas en neutrófilos y en las mayores de 48 horas generalmente predominan los linfocitos4,20. Hay reportes que afirman que la neutrofilia intersticial es más común en los pacientes con UVH, mientras que la eosinofilia es más frecuente en aquellos con UVN6,14.
Aquellos autores que se adhieren a criterios más estrictos para definir UV consideran que la leucocitoclasia y los depósitos de fibrina son las características más importantes para definir la entidad, puesto que representan signos directos de injuria vascular10. Sin embargo, otros han sugerido que los cambios histopatológicos presentes en la UV forman parte de un espectro. Así, en las lesiones menos severas se observa un infiltrado perivascular disperso; en el intermedio se encuentra un infiltrado perivascular denso con leucocitoclasia mínima pero sin depósitos de fibrina; y al final del espectro se encuentran los casos con leucocitoclasia franca y depósitos de fibrina3,11,21,22. Los cambios histopatológicos más severos generalmente se acompañan de síntomas sistémicos más graves.
Autores como Aboobaker y Lee han extendido la definición de UV para incluir pacientes con infiltrado perivascular predominantemente linfocítico13,23,24; sin embargo, esta posición no está totalmente aceptada25.
Inmunopatología
Más de la mitad de los pacientes con UV presentan inmunofluorescencia positiva que muestra depósitos de inmunoglobulinas, complemento y/o fibrinógeno en las paredes de los vasos sanguíneos. El depósito granular de complemento e inmunoglobulinas también se puede apreciar en la membrana basal de la piel22. Mehregan et al. observaron inmunorreactantes en las paredes de los vasos y/o en la membrana basal en el 79% de las lesiones cutáneas estudiadas5. La inmunofluorescencia positiva en la membrana basal en un paciente con UV hipocomplementémica sugiere el diagnóstico de LES6. Algunos autores sugieren que el compromiso renal se puede predecir de cierto modo por los depósitos de inmunoreactantes; en el estudio de Sánchez y colaboradores la inmunofluorescencia directa positiva en la unión dermoepidérmica se asoció con glomerulonefritis en el 70% de los pacientes26.
Patofisiología
Se considera que la UV representa una reacción de hipersensibilidad tipo III3. Clásicamente se ha postulado que al igual que lo que sucede con las demás vasculitis leucocitoclásticas, los complejos antígeno (Ag) anticuerpo (Ab) circulantes se forman inicialmente en la sangre y luego se depositan en las paredes de los vasos sanguíneos. El complemento es activado a través de la vía clásica, generando C3a y C5a. Estas anafilotoxinas inducen la degranulación de mastocitos y la síntesis de novo de citoquinas y quimoquinas, lo que lleva a un aumento de la permeabilidad vascular, quimiotaxis de neutrófilos y mayores depósitos de complejos inmunes. En el sitio de inflamación los neutrófilos liberan enzimas proteolíticas (colagenasa y elastasa) que exacerban la destrucción tisular y el edema2.
Kano y colaboradores, mediante la inducción de UV a través de ejercicio, proponen una secuencia de eventos para explicar cómo se desarrolla la entidad. El proceso inicia con el depósito de complejos inmunes que es seguido por una activación de mastocitos y liberación de factor de necrosis tumoral alfa (TNFα), influjo de eosinófilos y depósito de gránulos de proteínas eosinofílicas. Luego se presenta una activación persistente de mastocitos e influjo de neutrófilos con liberación de enzimas proteolíticas que finalmente conllevan al daño vascular. Los autores encontraron que los eosinófilos fueron las primeras células presentes en la lesión, seguidas por los neutrófilos, por lo que sugieren que el depósito extracelular extenso de gránulos de proteínas eosinofílicas (tres horas) que se presenta previamente a la llegada de los neutrófilos (10 a 24 horas) es un evento biológico clave en la patogénesis de la UV27.
Kano y colaboradores también reportaron una expresión intensa de TNFα, E selectina y molécula de adhesión intercelular 1 (ICAM-1) en las biopsias de lesiones tempranas27. Se postula que el TNFα liberado por los mastocitos puede aumentar la expresión de ICAM-1 facilitando la migración de los eosinófilos, y de E-selectina facilitando la migración de neutrófilos28,29.
Múltiples antígenos, tanto exógenos como endógenos, han sido implicados en la formación de anticuerpos que resultan en complejos inmunes que se depositan en los vasos sanguíneos de los pacientes con UV. Entre estos se encuentran agentes virales, medicamentos y la porción similar al colágeno del C1q. Sin embargo, en la mayoría de los casos este antígeno es desconocido2,10.
Los virus de la hepatitis B y la hepatitis C están entre los agentes infecciosos capaces de inducir la formación de estos complejos inmunes. Los antígenos de superficie de la hepatitis B y C se han aislado de los vasos afectados de pacientes con UV30.
Entre los antígenos endógenos se encuentra la región similar al colágeno del C1q8. Los anticuerpos contra este autoantígeno son moléculas IgG que se unen a la porción Fc del C1q, activando la vía clásica del complemento y causando hipocomplementemia8,21,31,32. Estos anticuerpos se encuentran en el 100% de los pacientes con síndrome de urticaria vasculítica hipocomplementémica (SUVH)33; sin embargo, no son exclusivos de esta patología, y también se han reportado en el 30% a 35% de pacientes con LES32 y hasta en el 83% de pacientes con LES y glomerulonefritis asociada34. Hay reportes en la literatura de pacientes con síndrome de Goodpasture35, hepatitis36 y vasculitis reumatoide, en ausencia de UV, con anticuerpos anti C1q10.
Aunque el rol de los anti C1q permanece desconocido en la patogénesis del LES y la UV2,4,10, se ha propuesto que estos anticuerpos son importantes en la patogénesis de la inflamación pulmonar que presentan los pacientes con SUVH. Se piensa que los anti C1q hacen reacción cruzada con la apoproteína del surfactante pulmonar que contiene una región semejante a la región similar al colágeno del C1q. En condiciones normales el surfactante regula la tensión alveolar superficial, por lo que se piensa que su deficiencia podría contribuir a la enfermedad pulmonar en pacientes con UV37. Hunt y colaboradores reportaron la coexistencia de capilaritis pulmonar y enfisema panancinar en un paciente con SUVH. Ellos postulan que la patogénesis de la enfermedad pulmonar obstructiva crónica (EPOC) en los pacientes con UV se relaciona con la VL que se presenta en los vasos pulmonares. Esta VL podría ser secundaria a la liberación de elastasa por los neutrófilos que conlleva a la destrucción del tejido pulmonar38.
Como se mencionó anteriormente, los anticuerpos anti C1q tienen una alta correlación con la glomerulonefritis proliferativa en los pacientes con LES (83%)34,39. Aunque es claro que los anticuerpos anti DNA de doble cadena (dsDNA) están implicados en la patogénesis de la glomerulonefritis en el LES, los anti C1q podrían corresponder a un epifenómeno4. Mannik y colaboradores proponen que los anti C1q contribuyen a la injuria glomerular al reaccionar con el C1q que está unido a los complejos inmunes formados por dsDNA-anti dsDNA y que esta unión podría hacer estos depósitos inmunes más solubles y/o más inflamatorios39. Algunos investigadores consideran que el posible rol de los anti C1q en la patogénesis de la enfermedad renal en los pacientes con LES podría extrapolarse a la patogénesis de la enfermedad renal en los pacientes con UV2,10.
Manifestaciones cutáneas
La UV clínicamente se caracteriza por presentar habones que duran más de 24 horas y frecuentemente pueden persistir hasta por tres a cuatro días. Las lesiones son recurrentes y se presentan por un periodo superior a seis semanas. En la mayoría de los casos son pruriginosas, y se pueden asociar a sensación de ardor y quemazón, aunque en algunos pacientes son asintomáticas3. Clásicamente se ha descrito que las lesiones dejan hiperpigmentación residual, pero en diferentes series esto sólo se ha encontrado en 25% a 35% de los pacientes5,14. Los habones pueden variar en forma y tamaño, pero algunos investigadores sugieren que las lesiones de UV son más pequeñas (0,5-1,5 cm) que las lesiones de urticaria alérgica4 (Ver Figuras 1 y 2). A diferencia de la púrpura palpable que tiene predilección por los miembros inferiores, los habones de UV pueden presentarse en cualquier parte del cuerpo. En algunos casos, la UV puede aparecer en sitios de presión, lo que la hace clínicamente indistinguible de la urticaria por presión3,10. Cuando la vasculitis afecta los capilares y vénulas postcapilares de la dermis profunda y el tejido celular subcutáneo, los pacientes con UV pueden presentar angioedema asociado, que se ha reportado hasta en el 42% de los casos5.
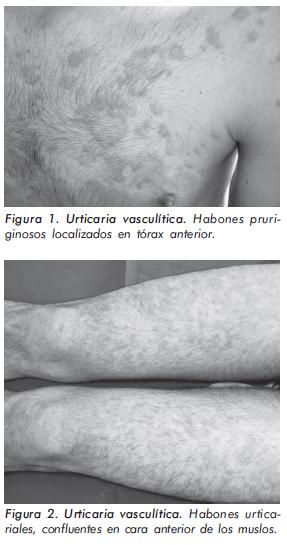
Manifestaciones cutáneas más raras asociadas con UV incluyen: lesiones similares al eritema multiforme, lesiones anulares, púrpura palpable, livedo reticularis, fenómeno de Raynaud y lesiones ampollosas3,8,33.
Se han propuesto diferentes métodos clínicos para ayudar a predecir si una lesión urticarial corresponde a una UV. Se sugiere que la presencia de glóbulos purpúricos a la dermatoscopia puede ser una clave diagnóstica que sugiere VL subyacente40. Igualmente la presencia de una mácula roja o marrón en el centro de un habón observado por diascopia sugiere la presencia de púrpura subyacente y puede ser usada como una perla diagnóstica para diferenciar la UV de la urticaria común41. Sin embargo, dado que las lesiones de UV pueden ser clínicamente indistinguibles de las lesiones de urticaria común, es importante recalcar que en un paciente que se presenta con más de cuatro a seis semanas de lesiones urticariales persistentes o recurrentes, es indispensable realizar una biopsia para esclarecer si se trata de una urticaria crónica de otra etiología o de una verdadera UV4.
Manifestaciones sistémicas
La UV puede clasificarse como UV primaria en cuyo caso corresponde un proceso de carácter idiopático y UV secundaria cuando se asocia a una enfermedad subyacente4. Según los niveles de complemento esta entidad puede subdividirse en urticaria vasculítica normocomplementémica (UVN) e hipocomplementémica (UVH)4,10,12. Las grandes series de pacientes con UV demuestran que la UVN es más común que la UVH5,6,14,26. Para la clasificación se sugiere la medición de C3, C4, CH50 y anti-C1q, al menos en tres ocasiones durante seis meses; si alguno de estos estudios muestra resultados anormales, el paciente debe ser clasificado como UVH12.
Se considera que los niveles de complemento son predictores de enfermedad sistémica. Múltiples grupos de investigadores han demostrado que los pacientes con UVH son mucho más propensos a presentar manifestaciones extracutáneas que los pacientes que no presentan activación del complemento5,6,14,26,33. El LES y la crioglobulinemia son la causa de las lesiones urticariales en la mayoría de pacientes con UVH, y aunque no se sabe exactamente cuál es la proporción de pacientes con UVH que cumplen con criterios diagnósticos de SUVH, se presume que este síndrome constituye menos del 5% de los casos. Por su parte, la mayoría de las UVN son de carácter idiopático y tienen pocas manifestaciones sistémicas; sin embargo, se han descrito pequeños números de pacientes con UVN asociada a gamapatía monoclonal, neoplasia, sensibilidad a la luz ultravioleta, exposición repetida al frío, esclerosis sistémica localizada y medicamentos4,12.
No hay parámetros clínicos precisos para hacer una distinción entre la UVN, la UVH y el SUVH. Todavía no es claro si estos subtipos de UV representan un espectro de la misma enfermedad o corresponden a desórdenes clínicos diferentes con patología similar. Investigadores con gran experiencia en el tema como Wisnieski sugieren que no hay transición de un subtipo de UV a otro4.
Las artralgias son la manifestación sistémica más común en los pacientes con UV y se presentan hasta en el 50% de los casos5. Estas son de carácter migratorio y generalmente aparecen de forma paralela con las lesiones cutáneas. Las articulaciones más afectadas en orden de frecuencia son: manos, codos, pies, tobillos y rodillas. La artritis es poco frecuente en los pacientes con UVN; sin embargo, puede estar presente hasta en el 50% de los pacientes con UVH26.
Aunque la artropatía de Jaccoud es más frecuente en los pacientes con LES, también puede presentarse en pacientes con UVH42,43. Hasta el momento se han reportado seis casos de UVH asociada a artropatía de Jaccoud y valvulopatía cardiaca, por lo que algunos recomiendan realizar ecocardiografía a estos pacientes44-47.
La manifestación pulmonar más frecuente en los pacientes con UV es la obstrucción de las vías aéreas que se puede presentar como asma o EPOC. Otras manifestaciones pulmonares menos frecuentes son: pleuritis, derrame pleural y hemoptisis48. En la serie de Mehregan et al.5 20% de los pacientes con UV presentaron compromiso pulmonar, mientras que en la serie de Davis et al.6 este compromiso se presentó en 17% de 24 pacientes con UVH y en 5% de 108 pacientes con UVN. A pesar de que la mayoría de pacientes con EPOC y UV son fumadores, la severidad del compromiso pulmonar excede a la esperada en fumadores sin UV. Además se han reportado varios casos de EPOC en pacientes jóvenes (30 a 40 años) con UV sin historia de tabaquismo, lo que hace pensar que la UV per se puede contribuir a la patogénesis de la enfermedad pulmonar. Aunque las complicaciones pulmonares pueden aparecer tardíamente, se considera que constituyen la principal causa de mortalidad en pacientes con UV48.
El compromiso renal se manifiesta con microhematuria y proteinuria en un 20% a 30% de pacientes con UVH49. Generalmente los pacientes con UVN no desarrollan compromiso renal26. La histología de las biopsias renales de estos pacientes presenta un amplio rango de hallazgos que pueden ser consistentes con glomerulonefritis proliferativa, vasculitis focal necrotizante, nefritis tubulointersticial, glomerulonefritis crescéntica y glomerulonefritis membranoproliferativa10. En ausencia de una enfermedad del colágeno de base, la progresión de la enfermedad renal en los pacientes con UV tiene un curso lento y raramente conlleva a enfermedad renal terminal3,49.
Las manifestaciones gastrointestinales se presentan en aproximadamente 17% a 30% de los pacientes con UV e incluyen: dolor abdominal, náuseas, vómito y diarrea26. La hemorragia gastrointestinal no se ha reportado en estos pacientes50.
El compromiso ocular ocurre en menos del 10% de los pacientes con UV y se manifiesta con epiescleritis, uveítis o conjuntivitis5,26,50. El compromiso cardíaco es raro, pero se han reportado casos de pericarditis recurrente asociada a taponamiento cardíaco37, derrame pericárdico51, y como se mencionó anteriormente, valvulopatías que pueden asociarse a artropatía de Jaccoud44-47. Aunque las complicaciones a nivel del sistema nervioso central son poco frecuentes, éstas incluyen: pseudotumor cerebri26,52, meningitis bacteriana53 y parálisis de pares craneanos54. Los pacientes con UV pueden presentar síntomas constitucionales como fiebre (10%), malestar y fatiga5. Otros hallazgos menos frecuentes son hepatomegalia, esplenomegalia50, linfadenopatías55, miositis56, atrofia del nervio óptico26 y mielitis transversa56.
Síndrome de urticaria vasculítica hipocomplementémica (SUVH)
El SUVH representa una forma más grave de UV descrita por McDuffie et al. en la Clínica Mayo en 19731. En 1982 Schwartz estableció los criterios preliminares mayores y menores para hacer el diagnóstico de SUVH48 (Ver Tabla 1).

Otras manifestaciones frecuentes en este síndrome que no están incluidas en los criterios diagnósticos son: angioedema, EPOC y varias manifestaciones neurológicas33. Aunque algunos pacientes con UV pueden tener hipocomplementemia, no todos cumplen los criterios diagnósticos del SUVH. Se dice entonces que estos pacientes tienen UVH pero no necesariamente SUVH10.
La identidad del SUVH como una enfermedad del tejido conectivo es debatida y dado que el SUVH comparte muchas características con el LES, algunos autores lo consideran un tipo inusual de LES. Davis et al. encontraron que el SUVH está presente en 7% a 8% de los pacientes con LES y que el 54% de los pacientes con SUVH cumplen con criterios de LES al seguirlos en el tiempo6. Existen varios reportes de casos en la literatura de pacientes que cumplen tanto criterios de clasificación de LES como de SUVH58,59.
Wisnieski ha reconocido el SUVH como una enfermedad autoinmune aparte, poco común, similar al LES. Considera que por definición todos los pacientes con este síndrome deben tener anticuerpos anti C1q detectados por ELISA. Según sus observaciones, los pacientes con SUVH activo raramente pueden tener niveles séricos normales de C3 y C4, pero siempre presentan niveles severamente deprimidos de C1q4,33.
Quienes defienden que el SUVH y el LES son entidades distintas, recalcan algunas diferencias diferencias entre estas dos patologías4,33. Entre estas se encuentran:
- La incidencia de angioedema es mucho mayor en el SUVH (50%) que en el LES.
- El EPOC, que se presenta hasta en el 50% de los pacientes con SUVH, y la inflamación del tracto uveal, que se presenta hasta en el 30% de los pacientes con SUVH, no son manifestaciones del LES.
- Todos los pacientes con SUVH tienen anticuerpos anti C1q asociados a lesiones de UV, pero raramente los pacientes con LES seropositivos para anti C1q desarrollan lesiones de UV, y por el contrario en el contexto del LES estos anticuerpos se correlacionan más con glomerulonefritis.
- A diferencia del LES en el cual casi el 100% de los pacientes tienen ANAs positivos, sólo el 50% a 60% de pacientes con SUVH tienen ANAs positivos en títulos moderados y en este último grupo de pacientes los anticuerpos anti dsDNA son transitorios e infrecuentes.
Independiente de los múltiples debates y discusiones al respecto, lo más probable es que la patogénesis del SUVH y el LES sea similar y que ambas entidades hagan parte de un mismo espectro de enfermedad autoinmune.
Factores causales y asociaciones
La mayoría de los casos de UV son de carácter idiopático3,10. Con menos frecuencia, especialmente en los casos de UVH, ésta se puede asociar a LES60,61, crioglobulinemia mixta esencial62 o síndrome de Sjögren63. Davis et al. estudiaron las diferencias entre los pacientes con UVH y UVN. De 132 pacientes con diagnóstico de UV confirmado por biopsia, 24 (18%) tenían complemento bajo y 13 (54%) cumplían con criterios de clasificación de LES. Los 108 pacientes restantes tenían complemento normal, y solo 3 (3%) cumplían con criterios de LES6. Alexander et al. reportaron que el 32% de 22 pacientes con síndrome de Sjögren tenían lesiones de UV60. La enfermedad del suero causa vasculitis debido al depósito de complejos inmunes, y frecuentemente se manifiesta con habones urticariales que a la histopatología pueden presentar venulitis dérmica e inflamación64.
Se han realizado múltiples asociaciones entre la UV y diferentes enfermedades sistémicas y exposiciones a medicamentos (ver Tabla 2); sin embargo, éstas son mucho más raras que las ya descritas y debido a la naturaleza anecdótica de los reportes, es casi imposible evaluar la relación causa efecto3,4,10.

Estudios adicionales en urticaria vasculítica
Cuando se sospecha una UV el primer paso a seguir es tomar una biopsia de las lesiones cutáneas que debe ser enviada para estudio con hematoxilina eosina e inmunofluorescencia directa3,4,10. Idealmente la muestra se debe tomar de una lesión reciente, y en ciertos casos es necesario tomar varias biopsias para confirmar el diagnóstico3,10. Aunque la definición de vasculitis ha variado en los diferentes estudios, nos adherimos a los criterios de los investigadores que consideran que la presencia de leucocitoclasia y destrucción del vaso con o sin depósitos fibrinoides son esenciales para hacer el diagnóstico de UV3,10.
Una vez se ha hecho el diagnóstico histológico de UV, todos los pacientes deben ser reevaluados para realizar una historia clínica completa y un examen físico cuidadoso, con el fin de descartar una enfermedad sistémica asociada4. No hay unos exámenes de rutina que se deban realizar a todos los pacientes con UV, y se recomienda que los paraclínicos se soliciten de acuerdo a los hallazgos encontrados en la historia clínica y el examen físico3,10. Algunos estudios básicos que se pueden solicitar inicialmente deben incluir: hemoleucograma completo con sedimentación, electrolitos, pruebas de función renal y hepática, niveles séricos de complemento (C1, C3, C4 y CH50), medición de complejos inmunes circulantes, inmunoglobulinas y crioglobulinas. Dependiendo de los hallazgos en la historia o los resultados de los paraclínicos iniciales, los pacientes pueden ser estudiados más a fondo con pruebas de función pulmonar, lavado broncoalveolar, rayos X osteoarticulares y ecocardiografía3,10,12.
La medición de ANAs, anticuerpos anti DNA, anti ENAs, anticuerpos antifosfolípidos, anticuerpos anti C1q, factor nefrítico de C3, serología para hepatitis B y C, el monotest para infección por virus Epstein-Barr y el test de Schirmer pueden ser exámenes útiles para investigar las causas y asociaciones de la UV en casos específicos3,10,12.
La clasificación de UV como normocomplementémica o hipocomplementémica requiere mediciones séricas de C1q, C3, C4 y CH50 en dos o tres ocasiones que deben realizarse en un periodo de observación de seis meses. Si cualquiera los niveles de complemento sérico están disminuidos en cualquier momento, debe considerarse que el paciente presenta una UVH4.
Las anormalidades más frecuentemente reportadas en las series más grandes de pacientes con UV incluyen eritrosedimentación elevada, hipocomplementemia (usualmente C3 y C4), ANAs positivos en títulos bajos y complejos inmunes circulantes5,6,22. Aunque la eritrosedimentación puede estar elevada hasta en el 75% de los pacientes con UV, este hallazgo es bastante inespecífico y no se ha asociado con mayor severidad o compromiso sistémico22,33.
Tratamiento
La UV es una enfermedad difícil de tratar. No existe una terapia universalmente efectiva, y la respuesta interindividual es muy variable3. Dado que no se han realizado estudios controlados aleatorizados que sustenten un tratamiento específico, se recomienda guiar el manejo de estos pacientes de acuerdo con la severidad del compromiso clínico (cutáneo vs. sistémico), la presencia o ausencia de enfermedad subyacente y el perfil de efectos secundarios de las distintas modalidades terapéuticas3,4,10.
Los antihistamínicos pueden ser útiles como monoterapia en casos muy leves que no estén asociados con enfermedad sistémica2,3,10,26,55. En la mayoría de los casos se utilizan en combinación con otros tratamientos como adyuvantes para controlar el prurito. No alteran el curso natural de la enfermedad puesto que no tienen actividad contra la inflamación desencadenada por el depósito de complejos inmunes2,10.
Los AINEs son frecuentemente utilizados en el manejo de la UV. La Indometacina es la más usada, y se han reportado tasas de respuesta hasta del 50% de los pacientes tratados con este medicamento5,13. Su uso está limitado por la intolerancia gastrointestinal2,3,10,26,55.
Los resultados de los casos de UV tratados con colchicina son variables, y aunque hay reportes que sugieren que es efectiva65,68, no todos los pacientes responden a este tratamiento5,69. Se piensa que actúa porque disminuye la migración de polimorfonucleares (PMN), estabiliza sus membranas lisosomales e inhibe su degranulación2,10.
Se han reportado múltiples casos de remisiones sostenidas en pacientes con UVH tratados con dapsona13,33,70-73. Por su amplio rango terapéutico, fácil administración, bajo costo y buena efectividad a largo plazo, algunos han considerado que la dapsona es la droga de elección en pacientes con UVH72. Otros autores consideran que los individuos con UV asociada a LES o cuadros similares al LES pueden tener mejor respuesta a este medicamento2. La dapsona actúa sobre los PMN, inhibiendo su sistema mieloperoxidasa, actividad lisosomal, generación de radicales libres, quimiotaxis y adherencia10.
La hidroxicloroquina es útil en el 50% de los pacientes con compromiso limitado a la piel55,74. Se piensa que actúa al inhibir la liberación de interleuquina 1 y enzimas lisosomales2,10. Tiene un efecto ahorrador de esteroides.
Los corticosteroides son efectivos en la mayoría de los pacientes con UV; sin embargo, su uso se ve limitado por el perfil de efectos secundarios a largo plazo2,3,5,10,12. Se deben reservar para los pacientes más comprometidos que no responden a otros medicamentos3. Su dosis depende de la severidad del cuadro clínico5,10. Generalmente los pacientes con UVH requieren esteroides sistémicos2,10,11,26,55. Aunque en algunos casos los cursos cortos son suficientes para manejar la UV, la mayoría de los pacientes presentan recaídas al suspender los esteroides, haciendo necesario su uso crónico2. La combinación con otros inmunosupresores como la azatioprina, dapsona, ciclofosfamida, ciclosporina y mofetil micofenolato, puede mejorar su eficacia y disminuir las dosis requeridas, reduciendo los efectos adversos2,3,10.
El metotrexate se ha utilizado como monoterapia o en combinación con otros medicamentos para el manejo de UV con tasas de respuesta variables12,75. Se describe un caso de exacerbación de UV luego de haber sido tratado con metotrexate76. En general no se considera como primera línea de tratamiento12.
El uso de azatioprina combinada con esteroides puede ser beneficioso en pacientes con UV y compromiso renal2,10,11.
La ciclofosfamida se reserva para los casos más graves y refractarios, generalmente en combinación con cortiscosteroides77.
La ciclosporina A puede ser útil en pacientes con SUVH4, incluso en casos refractarios al manejo con ciclofosfamida2. Puede disminuir la proteinuria en pacientes con UV y compromiso renal10,78. Además, se ha reportado mejoría del volumen espiratorio forzado en un segundo (VEF1) y de la capacidad de difusión alveolar de monóxido de carbono (DCLO) en pacientes con UV y compromiso pulmonar tratados con ciclosporina4.
El mofetil micofenolato ha sido efectivo como terapia de mantenimiento en pacientes con SUVH previamente tratados con ciclofosfamida y esteroides79,80.
La remoción de los complejos inmunes circulantes con plasmaféresis puede ser útil en casos de UV recalcitrante puesto que rápidamente provee de un beneficio temporal. Sin embargo, los pacientes recaen 24 horas después de suspender el tratamiento13,21.
El interferón alfa está indicado en el manejo de pacientes con infección por hepatitis C complicada por UV y crioglobulinemia81,82. Se puede utilizar en combinación con rivabirina para mejorar los resultados4.
Hay dos reportes en la literatura de pacientes con SUVH tratados exitosamente con altas dosis de inmunoglobulina intravenosa83,84. Se piensa que su mecanismo de acción en los pacientes con UV podría estar explicado por la capacidad de disminuir los depósitos de C3 a nivel de los vasos dérmicos84.
El rituximab es un anticuerpo monoclonal quimérico anti CD20 que induce la depleción de los linfocitos B in vivo85. Se reportó el caso de un paciente con SUVH, angioedema y LES, quien después de haber sido refractario a múltiples tratamientos respondió al manejo con rituximab59. Los autores proponen que el rituximab puede ser útil en el manejo del SUVH asociado a LES porque en estas patologías hay una disregulación de la producción de autoanticuerpos y producción de complejos inmunes, que está relacionada con una hiperactividad de las células B59.
Hay reportes de pacientes con UV tratados con calcio antagonistas, doxepina86 y pentoxifilina87. Sin embargo, muy probablemente estos medicamentos ofrecen pocos beneficios a los pacientes con SUVH o con otras enfermedades subyacentes tales como LES, síndrome de Sjögren y crioglobulinemia mixta4,10.
Pronóstico
La UV tiene un curso impredecible. Las lesiones pueden presentarse por algunas semanas o persistir por varios años3,10. En la serie de Mehregan que incluyó 72 pacientes, la duración promedio de la UV fue de tres años5. La UVN de carácter idiopático tiene un curso más benigno. Una de las más grandes series reportadas encontró que luego de un seguimiento de 12 años estos pacientes no desarrollaron otras manifestaciones sistémicas en el tiempo50. Los pacientes con UVH y LES asociado tienen un pronóstico menos favorable, siendo el EPOC50 y el edema laríngeo8 las causas más frecuentes de morbimortalidad. Es importante recalcar que aunque prácticamente todos los pacientes con UVH (idiopática o secundaria) tienen algún tipo de compromiso sistémico, pocos pacientes mueren como consecuencia directa de la UV2,26.
Agradecimientos fotografías
Doctora Ángela Londoño. Dermatóloga, docente, Universidad Pontificia Bolivariana, Medellín, Colombia.
Referencias
1. McDuffie FC, Sams WM Jr, Maldonado JE, et al. Hypocomplementemia with cutaneous vasculitis and arthritis. Possible immune complex syndrome. Mayo Clin Proc 1973; 48:340-348. [ Links ]
2. Venzor J, Lee WL, Huston DP. Urticarial vasculitis. Clin Rev Allergy Immunol 2002; 23:201-216. [ Links ]
3. Black AK. Urticarial vasculitis. Clin Dermatol 1999; 17:565-569. [ Links ]
4. Wisnieski JJ. Urticarial vasculitis. Curr Opin Rheumatol 2000; 12:24-31. [ Links ]
5. Mehregan DR, Hall MJ, Gibson LE. Urticarial vasculitis: a histopathologic and clinical review of 72 cases. J Am Acad Dermatol 1992; 26:441-448. [ Links ]
6. Davis MDP, Daoud MS, Kirby B, Gibson LE, Rogers RS. Clinicopathologic correlation of hypocomplementemic and normocomplementemic urticarial vasculitis. J Am Acad Dermatol 1998; 38:899-905. [ Links ]
7. Soter NA, Austen KF, Gigli I. Urticaria and arthralgias as manifestations of necrotizing angiitis (vasculitis). J Invest Dermatol 1974; 63:485-490. [ Links ]
8. Zeiss CR, Burch FX, Marder RJ, et al. A hypocomplementemic vasculitic urticarial syndrome: A report of four new cases and definition of the disease. Am J Med 1980; 68:867-875. [ Links ]
9. Agnello V, Ruddy S, Winchester RJ, et al. Hereditary C2 deficiency in systemic lupus erythematosus and acquired complement abnormalities in an unusual SLErelated syndrome. Birth Defects 1975; 11:312-317. [ Links ]
10. Davis MD, Brewer JD: Urticarial vasculitis and hypocomplementemic urticarial vasculitis syndrome. Immunol Allergy Clin North Am 2004, 24:183-213. [ Links ]
11. Monroe EW. Urticarial vasculitis: an updated review. J Am Acad Dermatol 1981; 5:88-95. [ Links ]
12. Brown NA, Carter JD. Urticarial vasculitis. Curr Rheumatol Rep. 2007; 9(4):312-319. [ Links ]
13. Aboobaker J, Greaves MW. Urticarial vasculitis. Clin Exp Dermatol 1986; 11:436-444. [ Links ]
14. Dincy CV, George R, Jacob M, Mathai E, Pulimood S, Eapen EP. Clinicopathologic profile of normocomplementemic and hypocomplementemic urticarial vasculitis: a study from South India. J Eur Acad Dermatol Venereol 2008; 22(7):789-794. [ Links ]
15. Tosoni C, Lodi-Rizzini F, Cinquini M, Pasolini G, Venturini M, Sinico RA, Calzavara-Pinton P. A reassessment of diagnostic criteria and treatment of idiopathic urticarial vasculitis: a retrospective study of 47 patients. Clin Exp Dermatol 2008; 2008 Aug 2. [Epub ahead of print] [ Links ]
16. Koch PE, Lazova R, Rosen JR, Antaya RJ. Urticarial vasculitis in an infant. Cutis 2008; 81(1):49-52. [ Links ]
17. Kaur S, Thami GP. Urticarial vasculitis in infancy. Indian J Dermatol Venereol Leprol 2003; 69(3):223- 224. [ Links ]
18. Soylu A, Kavukçu S, Uzuner N, Olgaç N, Karaman O, Ozer E. Systemic lupus erythematosus presenting with normocomplementemic urticarial vasculitis in a 4-yearold girl. Pediatr Int 2001; 43(4):420-422. [ Links ]
19. Soter NA. Chronic urticaria as a manifestation of necrotizing venulitis. N Engl J Med 1977; 296:1440- 1442. [ Links ]
20. Weedon D. Skin Pathology, 2nd ed. Churchill Livingstone, London, 2002: 232. [ Links ]
21. Russell Jones R, Bhogal B, Dash A, et al. Urticaria and vasculitis: A continuum of histopathological and immunopathological changes. Br J Dermatol 1983; 108:(110):139-149. [ Links ]
22. Berg RE, Kantor GR, Bergfeld WF. Urticarial vasculitis. Int J Dermatol 1988; 27:468-472. [ Links ]
23. Lee JS, Loh TH, Seow SC, Tan SH. Prolonged urticaria with purpura: the spectrum of clinical and histopathologic features in a prospective series of 22 patients exhibiting the clinical features of urticarial vasculitis. J Am Acad Dermatol 2007; 56:994-1005. [ Links ]
24. Kao NL, Zeitz HJ. Urticarial skin lesions and polymyositis due to lymphocytic vasculitis. West J Med 1995; 162:156-158. [ Links ]
25. Guitart J. "Lymphocytic vasculitis" is not urticarial vasculitis. J Am Acad Dermatol 2008; 59(2):353. [ Links ]
26. Sanchez NP, Winkelmann RK, Schroeter AL, Dicken CH. The clinical and histopathologic spectrums of urticarial vasculitis: study of forty cases. J Am Acad Dermatol 1982; 7:599-605. [ Links ]
27. Kano Y, Orihara M, Shiohara T. Cellular and molecular dynamics in exercise-induced urticarial vasculitis lesions. Arch Dermatol 1998; 134:62-67. [ Links ]
28. Chen K, Pittelkow MR, Su WP, et al. Recurrent cutaneous necrotizing eosinophilic vasculitis. Arch Dermatol 1994; 130:1159-1166. [ Links ]
29. Mehregan DR, Gibson LE. Pathophysiology of urticarial vasculitis. Arch Dermatol 1998; 134: 88-89. [ Links ]
30. Dienstag JL, Rhodes AR, Bhan AK, Dvorak AM, Mihm Jr MC, Wands JR. Urticaria associated with acute viral hepatitis type B: studies of pathogenesis. Ann Intern Med 1978; 89:34-40. [ Links ]
31. Wisnieski JJ, Jones SM. Comparison of autoantibody to the collagen-like region of C1q in hypocomplementemic urticarial vasculitis syndrome and systemic lupus erythematosus. J Immunol 1992; 148:1396-1403. [ Links ]
32. Wisnieski JJ, Jones SM. IgG autoantibodies to the collagen-like region of C1q in hypocomplementemic urticarial vasculitis syndrome, systemic lupus erythematosus, and 6 other musculoskeletal or rheumatic diseases. J Rheumatol 1992; 19:884-888. [ Links ]
33. Wisnieski JJ, Baer AN, Christensen J, Cupps TR, Flagg DN, Jones JV, Katzenstein PL, McMillen JJ, McFadden ER, Pick MA, Richmond GW, Simon S, Smith HR, Sontheimer RD, Trigg LB, Weldon D, Zone JJ. Hypocomplementemic urticarial vasculitis syndrome: clinical and serologic findings in 18 patients. Medicine 1995; 74:24-41. [ Links ]
34. Siegert CE, Daha MR, Westedt ML, van der Voort E, Breedveld F. IgG autoantibodies against C1q are correlated with nephritis, hypocomplementemia, and dsDNA antibodies in systemic lupus erythematosus. J Rheumatol 1991; 18:230-238. [ Links ]
35. Coremans IE, Daha MR, van der Voort EA, Muizert Y, Halma C, Breedveld FC. Antibodies against C1q in anti-glomerular basement membrane nephritis. Clin Exp Immunol 1992; 87: 256-260. [ Links ]
36. Lienesch DW, Sherman KE, Metzger A, et al. Anti-c1q antibodies in patients with chronic hepatitis C infection. Clin Exp Rheumatol 2006; 24:183-185. [ Links ]
37. Babjanians A, Chung-Park M, Wisnieski JJ. Recurrent pericarditis and cardiac tamponade in a patient with hypocomplementemic urticarial vasculitis syndrome. J Rheumatol 1991; 18:752-755. [ Links ]
38. Hunt DP, Weil R, Nicholson AG, Burke MM, Du Bois RM, Wells AU. Pulmonary capillaritis and its relationship to development of emphysema in hypocomplementemic urticarial vasculitis syndrome. Sarcoidosis Vasc Diffuse Lung Dis 2006; 23 (1): 70-72. [ Links ]
39. Mannik M, Wener MH. Deposition of antibodies to the collagen-like region of C1q in renal glomeruli of patients with proliferative lupus glomerulonephritis. Arthritis Rheum 1997; 40:1504-1511. [ Links ]
40. Vázquez-López F, Maldonado-Seral C, Soler-Sanchez T, Perez-Oliva N, Marghoob AA. Surface microscopy for discriminating between common urticaria and urticarial vasculitis. Rheumatology (Oxford) 2003; 42:1079-1082. [ Links ]
41. Dahl MV. Clinical pearl: diascopy helps diagnose urticarial vasculitis. J Am Acad Dermatol 1994; 30:481-482. [ Links ]
42. Sturgess AS, Littlejohn GO. Jaccoud's arthritis and panvasculitis in the hypocomplementemic urticarial vasculitis syndrome. J Rheumatol 1988; 15:858-861. [ Links ]
43. Ishikawa O, Miyachi Y, Watanabe H. Hypocomplementemic urticarial vasculitis associated with Jaccoud's syndrome. Br J Dermatol. 1997; 137(5): 804-807. [ Links ]
44. Palazzo E, Bourgeois P, Meyer O, De Bandt M, Kazatchkine M, Kahn M. Hypocomplementemic urticarial vasculitis syndrome, Jaccoud's syndrome, valvulopathy: a new syndrome combination. J Rheumatol 1993; 20:1236-1240. [ Links ]
45. Hong L, Wackers F, Dewar M, Kashgarian M, Askenase PW. Atypical fatal hypocomplementemic urticarial vasculitis with involvement of native and homograft aortic valves in an African American man. J Allergy Clin Immunol 2000; 106:1196-1198. [ Links ]
46. Chen HJ, Bloch KJ. Hypocomplementemic urticarial vasculitis, Jaccoud's arthropathy, valvular heart disease, and reversible tracheal stenosis: a surfeit of syndromes. J Rheumatol 2001; 28:383-386. [ Links ]
47. Amano H, Furuhata N, Tamura N, Tokano Y, Takasaki Y. Hypocomplementemic urticarial vasculitis with Jaccoud's arthropathy and valvular heart disease: case report and review of the literature. Lupus 2008 Sep;17(9):837-841. [ Links ]
48. Schwartz HR, McDuffie FC, Black LF, Schroeter AL, Conn DL. Hypocomplementemic urticarial vasculitis: association with chronic obstructive pulmonary disease. Mayo Clin Proc 1982; 57:231-238. [ Links ]
49. Kobayashi S, Nagase M, Hidaka S, Arai T, Ikegaya N, Hishida A, Honda N. Membranous nephropathy associated with hypocomplementemic urticarial vasculitis: report of two cases and a review of the literature. Nephron 1994; 66(1):1-7. [ Links ]
50. Soter NA. Urticarial vasculitis. In: Champion RH, Greaves MW, Kobza Black A, Pye RJ, editors. The urticarias. Edinburgh: Churchill Livingstone, 1985:141-148. [ Links ]
51. Jones MD, Tsou E, Lack E, Cupps TR. Pulmonary disease in systemic urticarial vasculitis: the role of bronchoalveolar lavage. Am J Med 1990; 88:431-434. [ Links ]
52. Ludivico CL, Myers AR, Maurer K. Hypocomplementemic urticarial vasculitis with glomerulonephritis and pseudotumor cerebri. Arthritis Rheum 1979; 22(9):1024-1028. [ Links ]
53. Alachkar H, Qasim F, Ahmad Y, Helbert M.Meningococcal meningitis in a patient with urticarial vasculitis: is there a link? J Clin Pathol 2007; 60(10):1160-1161. [ Links ]
54. Koul PA, Wahid A, Shah SU, Koul AN, Saleem SM. Hypocomplementemic urticarial vasculitis and lower cranial nerve palsies. J Assoc Physicians India 2000; 48:536-537. [ Links ]
55. Callen JP, Kalbfleisch S. Urticarial vasculitis: a report of nine cases and review of the literature. Br J Dermatol 1982; 107:87-94. [ Links ]
56. Chew GY, Gatenby PA. Inflammatory myositis complicating hypocomplementemic urticarial vasculitis despite on-going immunosuppression. Clin Rheumatol 2007; 26(8):1370-1372. [ Links ]
57. Bolla G, Disdier P, Verrot D, Swiader L, Andrac L, Harle JR, et al. Acute transverse mielitis and primary urticarial vasculitis. Clin Rheumatol 1998; 17:250-252. [ Links ]
58. Aydogan K, Karadogan SK, Adim SB, et al. Hypocomplementemic urticarial vasculitis: a rare presentation of systemic lupus erythematosus. Int J Dermatol 2006; 45:1057-1061. [ Links ]
59. Saigal K, Valencia IC, Cohen J, Kerdel FA. Hypocomplementemic urticarial vasculitis with angioedema, a rare presentation of systemic lupus erythematosus: rapid response to rituximab. J Am Acad Dermatol 2003; 49(5):S283-285. [ Links ]
60. O'Loughlin S, Schroeter AL, Jordon RE. Chronic urticaria- like lesions in systemic lupus erythematosus: a review of 12 cases. Arch Dermatol 1978; 114:879-883. [ Links ]
61. Provost TT, Zone JJ, Synkowski D, Maddison PJ, Reichlin M. Unusual cutaneous manifestations of systemic lupus erythematosus: I. Urticaria-like lesions. Correlation with clinical and serological abnormalities. J Invest Dermatol 1980; 75:495-499. [ Links ]
62. Kuniyuki S, Katoh H. Urticarial vasculitis with papular lesions in a patient with type C hepatitis and cryoglobulinemia. J Dermatol 1996; 123:279-283. [ Links ]
63. Alexander EL, Provost TT. Cutaneous manifestations of primary Sjögren's syndrome; a reflection of vasculitis and association with anti-Ro (SSA) antibodies. J Invest Dermatol 1983; 80:386-391. [ Links ]
64. Buhner D, Grant JA. Serum sickness. Dermatol Clin 1985; 3:107-117. [ Links ]
65. Werni R, Schwarz T, Gschnait F. Colchicine treatment of urticarial vasculitis. Dermatologica 1986; 172:36-40. [ Links ]
66. Wiles JC, Hansen RC, Lynch PJ. Urticarial vasculitis treated with colchicine. Arch Dermatol 1985; 121:802-805. [ Links ]
67. Callen JP. Colchicine is effective in controlling chronic cutaneous leukocytoclastic vasculitis. J Am Acad Dermatol 1985; 13:193-200. [ Links ]
68. Asherson RA, Buchanan N, Kenwright S, Fletcher CM, Hughes GR. The normocomplementemic urticarial vasculitis syndrome- report of a case and response to colchicine. Clin Exp Dermatol 1991; 16:424-427. [ Links ]
69. Muramatsu C, Tanabe E. Urticarial vasculitis: response to dapsone and colchicine. J Am Acad Dermatol 1985; 13:1055. [ Links ]
70. Matthews CN, Saihan EM, Warin RP. Urticaria-like lesions associated with systemic lupus erythematosus: response to dapsone. Br J Dermatol 1978; 99:455- 457. [ Links ]
71. Forston JS, Zone JJ, Hammond ME, Groggel GC. Hypocomplementemic urticarial vasculitis syndrome responsive to dapsone. J Am Acad Dermatol 1986; 15:1137-1142. [ Links ]
72. Eiser AR, Singh P, Shanies HM. Sustained dapsoneinduced remission of hypocomplementemic urticarial vasculitis–a case report. Angiology 1997; 48(11): 1019-1022. [ Links ]
73. Ruzicka T, Goerz G. Systemic lupus erythematosus and vasculitic urticaria. Effect of dapsone and complement levels. Dermatologica 1981; 162(3):203-205. [ Links ]
74. Lopez LR, Davis KC, Kohler PF, et al. The hypocomplementemic urticarial-vasculitis syndrome: Therapeutic response to hydroxychloroquine. J Allergy Clin Immunol 1984; 73:600-603. [ Links ]
75. Stack PS. Methotrexate for urticarial vasculitis. Ann Allergy. 1994; 72(1):36-38. [ Links ]
76. Borcea A, Greaves MW. Methotrexate-induced exacerbation of urticarial vasculitis: an unusual adverse reaction. Br J Dermatol 2000; 143(1):203-204. [ Links ]
77. Worm M, Muche M, Schulze P, Sterry W, Kolde G. Hypocomplementemic urticarial vasculitis: successful treatment with cyclophosphamide-dexamethasone pulse therapy. Br J Dermatol 1998; 139:704-707. [ Links ]
78. Soma J, Sato H, Ito S, Saito T. Nephrotic syndrome associated with hypocomplementemic urticarial vasculitis syndrome: successful treatment with cyclosporin A. Nephrol Dial Transplant 1999; 14:1753-1757. [ Links ]
79. Enríquez R, Sirvent AE, Amorós F, Pérez M, Matarredona J, Reyes A. Crescentic membranoproliferative glomerulonephritis and hypocomplementemic urticarial vasculitis. J Nephrol 2005;18(3): 318-322. [ Links ]
80. Worm M, Sterry W, Kolde G. Mycophenolate mofetil is effective for maintenance therapy of hypocomplementaemic urticarial vasculitis. Br J Dermatol 2000; 143(6): 1324. [ Links ]
81. Hamid S, Cruz Jr PD, Lee WM. Urticarial vasculitis caused by hepatitis C virus infection: response to interferon alpha therapy. J Am Acad Dermatol 1998; 39:278-280. [ Links ]
82. Misiani R, Bellavita P, Fenili D, Vicari O, Marchesi D, Sironi PL, et al. Interferon alfa-2a therapy in cryoglobulinemia associated with hepatitis C virus. N Engl J Med 1994; 330:751-756. [ Links ]
83. Staubach-Renz P, von Stebut E, Bräuninger W, Maurer M, Steinbrink K. Hypocomplementemic urticarial vasculitis syndrome. Successful therapy with intravenous immunoglobulin Hautarzt 2007; 58(8):693-697. [ Links ]
84. Shah D, Rowbottom AW, Thomas CL, Cumber P, Chowdhury MM. Hypocomplementaemic urticarial vasculitis associated with non-Hodgkin lymphoma and treatment with intravenous immunoglobulin. Br J Dermatol 2007;157(2):392-393. [ Links ]
85. Carr DR, Heffernan MP. Off-label uses of rituximab in dermatology. Dermatol Ther 2007; 20(4):277-287. [ Links ]
86. Kelkar PS, Butterfield JH, Kalaaji AN. Urticarial vasculitis with asymptomatic chronic hepatitis C infection: response to doxepin, interferon-alpha, and ribavirin. J Clin Gastroenterol 2002; 35:281-282. [ Links ]
87. Nurnberg W, Grabbe J, Czarnetzki BM. Urticarial vasculitis syndrome effectively treated with dapsone and pentoxifylline. Acta Derm Venereol 1995; 75: 54-56. [ Links ]

