










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Reumatología
Print version ISSN 0121-8123
Rev.Colomb.Reumatol. vol.16 no.3 Bogotá JulySep. 2009
PRESENTACIÓN DE CASOS
Pulmonary Complications of idiopathic inflammatory myopathies: a case with myopathy of the diaphragm
Mauricio Restrepo Escobar1, Luis Alonso González Naranjo1, Adelis Enrique Pantoja
Márquez2, Luis Alberto Ramírez Gómez1, Gloria María Vásquez Duque3
1 MD, Internista, Reumatólogo, Profesor Sección de Reumatología, Facultad de Medicina, Universidad de Antioquia, Hospital Universitario San Vicente de Paúl. Medellín, Colombia.
2 MD, Internista, Residente de Reumatología. Universidad de Antioquia, Hospital Universitario San Vicente de Paúl. Medellín, Colombia.
3 MD, PhD, Internista, Reumatóloga, Inmunóloga, Profesora Sección de Reumatología, Facultad de Medicina, Universidad de Antioquia, Hospital Universitario San Vicente de Paúl. Medellín, Colombia.
Recibido: Junio 18 de 2009 Aceptado: Agosto 28 de 2009
Resumen
Las miopatías inflamatorias idiopáticas son el mayor grupo de miopatías adquiridas. Con base en los hallazgos clínicos, histopatológicos, inmunológicos y demográficos pueden ser diferenciadas en tres diferentes subgrupos: dermatomiositis, polimiositis y miositis por cuerpos de inclusión. El compromiso pulmonar es cada vez más reconocido como una complicación grave y una causa frecuente de morbilidad y mortalidad en estas enfermedades. Existen tres categorías de complicaciones pulmonares en miositis inflamatoria: neumonía por aspiración, hipoventilación y enfermedad pulmonar intersticial. La falla respiratoria causada por hipoventilación es una complicación poco común de las miopatías inflamatorias, la cual se presenta en pacientes con debilidad muscular generalizada e inflamación de los músculos respiratorios de inspiración y espiración. La debilidad del diafragma es probablemente una manifestación frecuente e ignorada en las miopatías inflamatorias. Reportamos el caso de una mujer con miopatía inflamatoria en quien los hallazgos de la electromiografía de músculos diafragmas demostraron compromiso inflamatorio de este músculo.
Palabras clave: miopatías inflamatorias, polimiositis, dermatomiositis, miopatía del diafragma.
Summary
The idiopathic inflammatory myopathies are the largest group of acquired myopathies. On the basis of clinical, histopathological, immunological and demographic features, they can be differentiated into three distinct subsets: dermatomyositis, polymyositis and inclusion-body myositis. Pulmonary involvement is increasingly recognized to be a serious complication and a common cause of morbidity and mortality in these diseases. There are three categories of pulmonary complications in inflammatory myositis: aspiration pneumonia, hypoventilation, and interstitial lung disease. Respiratory failure caused by hypoventilation is considered to be an uncommon complication of inflammatory myositis which occurs in patients with severe generalized muscle weakness and inflammation of inspiratory and expiratory respiratory muscles. Diaphragm weakness is frequent and probably overlooked in inflammatory myopathies. We report the case of a woman with inflammatory myopathy in whom the findings of the diaphragm muscles electromyography demonstrated inflammatory involvement of this muscle.
Key words: inflammatory myopathies, polymyositis, dermatomyositis, myopathy of the diaphragm.
Introducción
Las miopatías inflamatorias idiopáticas conforman el grupo más grande de miopatías adquiridas. Estas enfermedades requieren de un diagnóstico rápido para instaurar un tratamiento oportuno. Debido a la naturaleza sistémica de las mismas deben descartarse complicaciones cutáneas, musculoesqueléticas, vasculares, gastrointestinales, cardiacas y pulmonares que pueden poner en riesgo la vida del paciente. Presentamos el caso de una paciente con polimiositis y síntomas y signos sutiles de afectación respiratoria en quien documentamos franca miopatía diafragmática y bajos volúmenes pulmonares.
Reporte de un caso
Mujer afro-colombiana de 30 años procedente de Quibdó, Chocó, con antecedente de anemia crónica quien ingresó al servicio de Neurología por cuadro clínico de año y medio de evolución de pérdida de 22 kilogramos de peso, debilidad muscular proximal en cinturas escapular y pélvica en el último año, la cual había sido más incapacitante en los últimos cuatro meses impidiendo la deambulación y limitando el autocuidado. No se informaron síntomas mucocutáneos, gastrointestinales o articulares, ni síntomas sugestivos de oftalmoplejía. En los últimos días ha presentado disnea intermitente, tos y estornudos inefectivos de baja tonalidad. Negaba la presencia de disfonía, disfagia, regurgitación nasal o tos con los alimentos. Durante la evaluación por el Servicio de Reumatología se encontró con cefaloparesia, atrofia muscular global, debilidad muscular de 2/5 proximal y 5/5 distal, con gran dificultad para mantenerse de pie y deambular. No se encontró eritema en heliotropo ni pápulas de Gottron. Sus niveles de creatina quinasa total oscilaron entre 21.748 y 35.782 U/L, con anticuerpos antinucleares por IFI de 1:80 (patrón moteado), y ENA negativos. No estuvieron disponibles autoanticuerpos específicos para miopatías inflamatorias. La electromiografía demostró signos de inestabilidad de membrana con potenciales polifásicos de corta duración en músculos de extremidades. Una electromiografía de músculos diafragmas demostró abundantes signos de inestabilidad de membrana, con descargas repetitivas complejas y potenciales de acción polifásicos, con baja amplitud y corta duración compatibles con alteración de la fibra muscular (Figura 1). Una tomografía de alta resolución del tórax fue normal, pero una espirometría reveló bajos volúmenes y flujos pulmonares. La ecocardiografía fue normal. En la biopsia de músculo deltoides se demostró haces de músculo estriado con variaciones en el tamaño y forma de sus fibras, con zonas de necrosis y vacuolización de estas, acompañado de infiltrado inflamatorio linfoplasmocitario y macrófagos, compatible con inflamación muscular crónica. La búsqueda de procesos infecciosos o neoplásicos fue negativa. Iniciamos manejo con un pulso diario de metilprednisolona por tres días seguidos de prednisolona 1 mg/kg/día en dosis fraccionada, 15 mg de metotrexate semanales. La paciente fue dada de alta con una mejoría clínica y de las pruebas de laboratorio satisfactorias, para continuar su seguimiento por la consulta externa del servicio de reumatología.
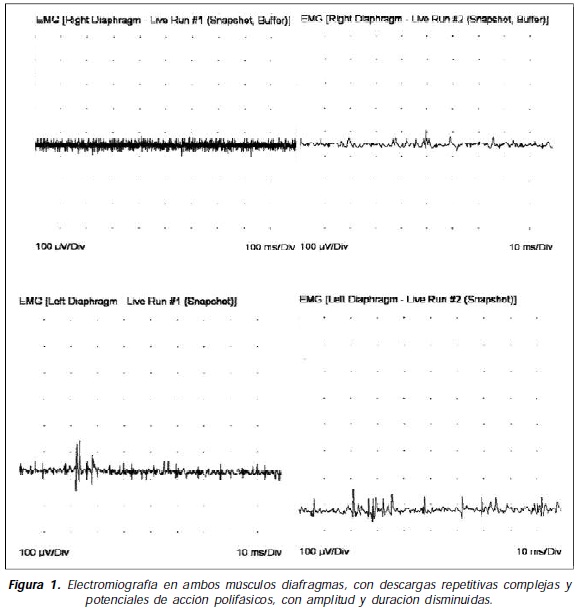
Discusión
Las miopatías inflamatorias idiopáticas son un grupo heterogéneo de enfermedades adquiridas del músculo esquelético, y constituyen el grupo más grande de miopatías adquiridas y potencialmente reversibles1. Este grupo de desórdenes se caracteriza por debilidad muscular proximal e inflamación no supurativa del músculo esquelético, frecuentemente acompañadas por manifestaciones extramusculares2.
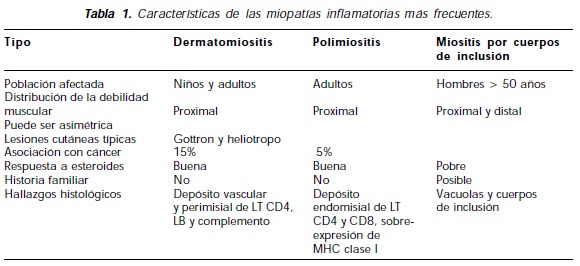
La polimiositis (PM), la dermatomiositis (DM) y la miositis por cuerpos de inclusión (MCI) son las tres principales categorías de las miopatías inflamatorias idiopáticas. Aunque ellas pueden compartir algunas similitudes, presentan características clínicas, histopatológicas, inmunológicas y patogénicas que las diferencian. La PM y la MCI son enfermedades mediadas por células T mientras que la DM está caracterizada por una microangiopatía mediada por el complemento. Es fundamental distinguir entre estos tres desórdenes ya que ellos difieren en el pronóstico y la respuesta al tratamiento3 (Tabla 1).
Los criterios utilizados más frecuentemente para el diagnóstico y la clasificación de las miopatías inflamatorias son aquellos propuestos por Bohan y Peter (Tabla 2). Estos criterios incluyen varios fenotipos diferentes de pacientes con miositis, lo cual indica que posiblemente existan diferentes etiologías, mecanismos patogénicos y pronósticos. Existen nuevas propuestas de clasificación que utilizan avances en la inmunohistopatología y los autoanticuerpos específicos de miositis, aunque carecen todavía de validación y aceptación global4.

La mayoría de los pacientes con miopatías inflamatorias presentan debilidad muscular como su síntoma principal de presentación. La debilidad muscular en estos pacientes tiene una presentación generalmente subaguda de predominio proximal y típicamente simétrica. En la miositis por cuerpos de inclusión la debilidad puede ser asimétrica y tempranamente afecta también musculatura distal. Pueden presentarse mialgias leves pero en general el dolor no es un síntoma típico en estas enfermedades5.
Todos los músculos pueden verse afectados aunque comúnmente se respetan los músculos faciales. La afectación de los músculos de faringe y el tercio superior del esófago puede manifestarse con voz nasal, regurgitación, tos de bajo tono, disfagia o aspiración crónica, complicando el curso de la enfermedad y constituyéndose en marcador de pobre pronóstico1.
De manera interesante, existen pacientes con miopatías inflamatorias quienes no presentan síntomas de debilidad muscular ni alteración de las enzimas de origen muscular. Estos pacientes reciben el diagnóstico de dermatomiositis amiopática porque presentan alteraciones cutáneas características de la enfermedad (confirmado o no con biopsia) pero no muestran signos o síntomas de miopatía. Algunos autores argumentan que al realizar biopsias musculares a estos pacientes se puede demostrar también algún grado de inflamación muscular5. Un creciente número de reportes, principalmente de pacientes asiáticos, informan que estos pacientes muestran mayor frecuencia de complicaciones pulmonares como neumonitis intersticial y neumomediastino/neumotórax o enfisema subcutáneo, a veces con muy pobre respuesta a la terapia6.
Existen muchas manifestaciones extramusculares de las miopatías inflamatorias, lo que indica su naturaleza sistémica. En la piel de los pacientes con dermatomiositis se encuentran lesiones típicas como el eritema en heliotropo y las pápulas de Gottron, así como también fotosensibilidad, signo del chal, signo de la V del cuello y poiquiloderma. En la piel también puede notarse la presencia de induraciones subcutáneas dolorosas o úlceras, ambas ocasionadas por calcinosis, así como la presencia de vasculitis y lipodistrofia los cuales son hallazgos característicos de la dermatomiositis juvenil. Un subgrupo de pacientes con polimiositis o dermatomiositis presentan un complejo sindromático caracterizado por fenómeno de Raynaud, artritis, fiebre, neumonitis intersticial, y autoanticuerpos contra algunas de las sintetasas del RNA de transferencia, denominándose como síndrome antisintetasa. En las manos de estos pacientes se pueden observar también lesiones cutáneas de aspecto sucio y con fisuras denominadas como "manos de mecánico"2,6,7.
El músculo cardíaco también puede afectarse aunque con baja frecuencia. Pueden ocurrir arritmias, falla cardíaca congestiva o cardiopatía dilatada. El desarrollo de miocarditis ha sido relacionado en algunos reportes con la presencia del autoanticuerpo anti-partícula de reconocimiento de señal. Cuando ha ocurrido hipertensión pulmonar, por mecanismos diversos como se verá más adelante, puede llegar a presentarse falla cardiaca congestiva por corpulmonale2.
Otras manifestaciones extramusculares de las miopatías inflamatorias incluyen alteraciones gastrointestinales, síntomas generales y constitucionales, artralgias/artritis, y manifestaciones pulmonares diversas. En ocasiones las manifestaciones extramusculares pueden ofrecer gran morbilidad para los pacientes más que el mismo músculo, y a veces la respuesta a la terapia puede ser discreta y sin correlación con el curso de la miopatía2.
Las complicaciones pulmonares de la PM y la DM pueden clasificarse en varios grupos; sin embargo, los tres principales tipos de complicaciones pulmonares en las miopatías inflamatorias son: la enfermedad pulmonar intersticial (EPI), la neumonía por aspiración y la hipoventilación. Aunque la EPI es la complicación más frecuente y devastadora, las otras manifestaciones pulmonares secundarias pueden complicar la evolución clínica de estos pacientes con consecuencias graves (Tabla 3). A pesar de que las complicaciones pulmonares se constituyen en la principal causa de morbimortalidad en los pacientes con miopatías inflamatorias, existen escasos reportes en la literatura y en general se enfocan en la enfermedad pulmonar intersticial8,9.

La incidencia reportada de EPI en la PM y la DM varía entre un 5% y un 65% dependiendo de los criterios utilizados para su definición o detección en cada estudio. Se ha observado un incremento en las frecuencias reportadas debido al uso más temprano de métodos diagnósticos más sensibles como las pruebas de función pulmonar y la tomografía axial computarizada de tórax alta resolución (TACAR) en los pacientes con miopatía inflamatoria tal como ha sucedido con otras enfermedades del colágeno10,11.
La expresión clínica de la EPI en pacientes con PM o DM puede variar desde estar completamente asintomático hasta disnea grave rápidamente progresiva con insuficiencia ventilatoria y desenlace fatal. Los pacientes pueden presentarse con tres patrones diferentes: con síntomas de aparición aguda, con síntomas crónicos lentamente progresivos, o sin síntomas y con hallazgos anormales en las imágenes o las pruebas de función pulmonar. La tos y la disnea son los síntomas más frecuentemente reportados, pero ellos cuentan con baja sensibilidad y especificidad para el diagnóstico de EPI. También es frecuente el hallazgo de crépitos basales típicamente descritos como en velcro10.
Deben tenerse presentes e investigar otros predictores de EPI tal como la presencia de anticuerpos anti-aminoacil tRNA sintetasa, principalmente el anti-histidil o anti-Jo-1, ya que es el más frecuentemente hallado. Del total de pacientes con miopatías inflamatorias aproximadamente el 20% tienen anticuerpos anti-Jo-1. Si se toman aquellos con EPI por miopatía inflamatoria la mitad tendrán anti-Jo-1. De todos los pacientes quienes tienen anti-Jo-1, más del 70% presentarán EPI. Se han propuesto otros biomarcadores de EPI pero tienen poca disponibilidad clínica y no han sido validados completamente12.
El diagnóstico de la EPI inicia desde la sospecha clínica; sin embargo, debido a que los síntomas y signos carecen de un rendimiento diagnóstico adecuado, los exámenes más útiles son las pruebas de función pulmonar, las cuales típicamente muestran un defecto ventilatorio restrictivo. La espirometría puede mostrar disminución de la capacidad pulmonar total, la capacidad funcional residual, el volumen residual, el volumen espiratorio forzado en un segundo (VEF1), y la capacidad vital forzada (CVF), pero con una relación VEF1/CVF normal o aumentada, y una disminución de la capacidad de difusión para el monóxido de carbono. La prueba más sensible es la capacidad de difusión de monóxido de carbono, pero no es específica de EPI; bajos niveles en la capacidad de difusión de monóxido de carbono (< 45%) han sido asociados con un pobre pronóstico en pacientes con miopatía inflamatoria y EPI13,14. Las placas de tórax pueden ofrecer información útil en casos avanzados de EPI o para descartar complicaciones como infección o neumotórax, pero pueden ser normales y de poca utilidad en las etapas tempranas de la EPI. La TACAR ofrece un mucho mejor rendimiento diagnóstico por lo cual es ampliamente utilizada en la actualidad para esta indicación. Aunque no es algo absoluto, una TACAR con buena técnica y en manos de un radiólogo experto puede informar la proporción relativa de imágenes sugestivas de inflamación activa frente a imágenes sugestivas de enfermedad fibrótica residual (vidrio esmerilado vs. panal de abejas). También es posible cuantificar con gran precisión la extensión y la gravedad de la afectación pulmonar, aportando información útil al momento del diagnóstico y para el pronóstico y seguimiento del paciente. Con un entrenamiento adecuado, un radiólogo puede aproximarse al subtipo (o subtipos) histológicos más probables, y su posible respuesta a la terapia8-10,15.
Aunque la biopsia pulmonar es el estándar de oro para el diagnóstico y la clasificación de la EPI, no se realiza en forma rutinaria debido al elevado riesgo de complicaciones en estos pacientes con alteración ventilatoria e intersticial de base. En caso de llevarse a cabo se requiere realizar una biopsia pulmonar a cielo abierto, y la lectura por parte de un patólogo experto en la compleja clasificación de la EPI8-10.
Al igual que en otras enfermedades del colágeno que pueden producir afectación pulmonar, en las miopatías inflamatorias se ha estudiado el lavado broncoalveolar para determinar el perfil celular inflamatorio, cuantificar biomarcadores, y descartar otras causas dentro del diagnóstico diferencial como infecciones, sarcoidosis o neumonitis por medicamentos. El papel del lavado broncoalveolar en la evaluación de la actividad inflamatoria y como guía del tratamiento todavía no ha sido validado ni ampliamente aceptado8,9.
Debido a que existen diferentes patrones histopatológicos de EPI con grados variables de respuesta a la terapia inmunosupresora se presume que existan diversos mecanismos patogénicos relacionados con estas complicaciones. Se han reportado diferentes patrones histopatológicos dentro de los cuales se incluyen la bronquiolitis obliterante con neumonía organizada, el daño alveolar difuso, la neumonía intersticial inespecífica, y la neumonía intersticial usual. El patrón de neumonía intersticial inespecífica ha sido informado como el más frecuente en varios reportes. No es infrecuente encontrar más de un patrón de enfermedad en una misma biopsia, lo cual dificulta la interpretación de una biopsia y la generación de recomendaciones sobre el uso rutinario de la biopsia pulmonar8-10.
Dentro del estudio del paciente con miopatía inflamatoria y EPI se han encontrado como factores de mal pronóstico la presentación como neumonía intersticial aguda, una capacidad de difusión de monóxido de carbono inicial menor del 45%, alveolitis neutrofílica y características histopatológicas de neumonía intersticial usual. Se considera en general que un patrón reticulado observado en una TACAR se correlaciona con el hallazgo histológico de fibrosis y pobre respuesta a la terapia, y por el contrario un patrón de vidrio esmerilado correlaciona con enfermedad inflamatoria potencialmente reversible con un mejor pronóstico. Algunos reportes sugieren un curso agresivo y un pobre pronóstico debido a EPI en pacientes con dermatomiositis amiopática16-20.
No se conoce el tratamiento óptimo para la EPI ocasionada por miopatía inflamatoria. La relativa rareza de la enfermedad y la heterogeneidad de su expresión clínica y manifestaciones pulmonares dificultan la realización de ensayos clínicos controlados. La información disponible basada en pequeñas series de casos señala a la terapia con glucocorticoides como la primera línea de manejo, como es el caso de los pacientes con miopatía inflamatoria sin EPI. Debido a que usualmente los esteroides son insuficientes para sostener una respuesta duradera, y que su uso prolongado a altas dosis aumenta la posibilidad de efectos adversos, generalmente se adiciona desde el principio otro agente inmunosupresor. Existen reportes favorables con ciclofosfamida, ciclosporina A, azatioprina, metotrexate y tacrolimus9,10,14.
Con respecto a otras manifestaciones pulmonares de las miopatías inflamatorias ellas se consideran secundarias a la enfermedad muscular o como complicaciones de la enfermedad o el mismo tratamiento. En un estudio el 17.3% de los pacientes presentó neumonía por aspiración. Se ha estimado que hasta el 50% de los pacientes con miopatías inflamatorias pueden presentar disfagia orofaríngea y regurgitación, ocasionadas por la disfunción del músculo estriado de la faringe y el esófago superior. Existe mayor riesgo cuando la afectación muscular o cutánea son extensas8,9.
La frecuente aspiración y la inmunosupresión intensa, así como la disminución de mecanismos de defensa como la tos, propicia la infección por gérmenes oportunistas. Esta complicación posee varios factores agravantes tales como la expresión clínica atípica, la confusión con neumonitis intersticial y la pobre reserva cardiopulmonar del paciente con miopatía8,9.
La falla cardiaca congestiva muestra una frecuencia baja de presentación, pero puede complicar el diagnóstico diferencial y la evolución clínica. Se han reportado casos de miocarditis relacionados con el autoanticuerpo anti-partícula de reconocimiento de señal. Puede ocurrir tanto falla cardiaca global como falla cardiaca derecha secundaria a hipertensión pulmonar, la cual a su vez puede tener distintas causas (vasculopatía primaria, embolismo pulmonar recurrente, o debido a hipoxia crónica por la neumonitis intersticial/ fibrosis pulmonar). La manifestación de la falla cardiaca con disnea e infiltrados difusos obliga a diferenciarla de infección pulmonar o de EPI. Se recomienda una evaluación ecocardiográfica periódica9.
La hipoventilación ocasionada por debilidad de los músculos de la ventilación parece ser una complicación rara, con informes que oscilan desde menos de 5% hasta el 21,8% de los pacientes. Es posible que esta complicación sea subdiagnosticada por su poca manifestación clínica, y por la dificultad para su demostración electromiográfica o histopatológica. Parece ser mucho más probable cuando existe afectación muscular grave y generalizada. Puede afectar músculos tanto inspiratorios como espiratorios. La hipoventilación puede generar disminución del reflejo protector de la tos y acumulación de moco, los cuales aumentan la posibilidad de atelectasias o neumonía8,9.
En la espirometría de los pacientes con hipoventilación también se puede observar un patrón restrictivo como en los pacientes con EPI, pero a diferencia de estos últimos en los primeros aumenta el volumen residual y se encuentran disminuidas las presiones inspiratorias y espiratorias máximas (PIM/PEM). Se ha propuesto como criterios de alarma para el desarrollo de falla ventilatoria unas PIM/PEM menores del 30% de lo predicho, o una capacidad vital menor del 50% de lo predicho. La placa de tórax de estos pacientes puede demostrar pulmones pequeños, hemidiafragmas elevados y atelectasias basales13.
Finalmente, el desarrollo espontáneo de neumomediastino, neumotórax o enfisema subcutáneo son manifestaciones raras pero con varios reportes en la literatura. Existen controversias respecto a su origen y a su manejo. Se ha postulado que pueden deberse a la ruptura de bulas subpleurales, lo cual puede estar facilitado por la presencia de tejidos debilitados por la neumonitis más el uso de esteroides, o incluso que pueden representar un fenómeno vasculítico asociado con la presencia de úlceras cutáneas en extremidades. El pronóstico en general no parece ser malo, y respecto al manejo se ha recomendado solo observar, utilizar oxígeno en altas concentraciones o un tubo a tórax, o incluso aumento o disminución de la inmunosupresión. También se han publicado casos similares en pacientes con otras enfermedades deltejido conectivo6,9,19,21,22.
Conclusiones
Las miopatías inflamatorias idiopáticas son enfermedades potencialmente reversibles con el manejo inmunosupresor y a pesar de su baja incidencia es importante diferenciarlas de otras miopatías que pueden presentarse clínicamente igual, para prevenir daño irreversible y potenciales complicaciones. Las complicaciones pulmonares primarias y secundarias de las miopatías inflamatorias son frecuentes y ocasionan gran morbilidad y mortalidad.
Estas complicaciones que pueden preceder, ser concomitantes o aparecer después de la miositis, requieren un bajo umbral diagnóstico por la presentación insidiosa y a veces asintomática. El diagnóstico oportuno y temprano es necesario para definir manejo y pronóstico y requiere de una tamización universal. Dentro del enfoque diagnóstico siempre deberán considerarse las posibilidades de infección, enfermedad pulmonar intersticial por la miopatía o por medicamentos, o edema pulmonar.
Faltan estudios de validación de las técnicas diagnósticas para el enfoque de la EPI en las miopatías inflamatorias, y no existen estudios clínicos controlados sobre el manejo de la misma. La historia natural del compromiso pulmonar intersticial se ve alterada por el uso casi universal de esteroides y otros inmunosupresores.
El mayor número de reportes de afectación respiratoria por miopatías inflamatorias obedece a neumopatía intersticial, siendo poco frecuente el reporte de debilidad de los músculos de la respiración, debido posiblemente al subdiagnóstico de esta complicación por los síntomas y signos clínicos sutiles. Sin embargo, la progresión de estas alteraciones puede predisponer a neumonías por aspiración o franca falla ventilatoria hipercápnica, por lo cual se requiere alto índice de sospecha para su detección temprana, manejo oportuno y monitorización adecuada.
La entidad clínica conocida como DM amiopática parece acompañarse de mayor frecuencia de complicaciones pulmonares, usualmente de carácter grave y curso refractario al manejo. Dentro de las manifestaciones pulmonares que parecen hallarse más frecuentemente en este subgrupo de pacientes sobresalen la EPI y el desarrollo de neumomediastino, neumotórax o enfisema subcutáneo espontáneos.
Referencias
1. Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet 2003;362:971-982. [ Links ]
2. Yazici Y, Kagen LJ. Clinical presentation of the idiopathic inflammatory myopathies. Rheum Dis Clin N Am 2002;28:823-832. [ Links ]
3. Briani C, Doria A, Sarni-Puttini P, Dalakas MC. A.-P. Update on idiopathic inflammatory myopathies. Autoimmunity 2006;39:161-170. [ Links ]
4. Mastaglia FL, Garlepp MJ, Phillips BA, Zilko PJ. Inflammatory Myopathies: Clinical, Diagnostic and Therapeutic Aspects. Muscle Nerve 2003;27:407-425. [ Links ]
5. Christopher-Stine L, Plotz PH. Adult inflammatory myopathies. Best Pract Res Clin Rheum 2004;18:331-344. [ Links ]
6. Nagaraju K, Lundberg IE. Inflammatory Diseases of Muscle and Other Myopathies. Kelley's Textbook of Rheumatology, 8th ed. W. B. Saunders Company 2008. [ Links ]
7. Sontheimer RD. Dermatomyositis: an overview of recent progress with emphasis on dermatologic aspects. Dermatol Clin 2002;20:387-408. [ Links ]
8. Fathi M, Lundberg IE, Tornling G. Pulmonary complications of polymiositis and dermatomyositis. Semin Respir Crit Care Med 2007;28:451-458. [ Links ]
9. Ascherman DP. Pulmonary Complications of Inflammatory Myopathy. Current Rheumatology Reports 2002;4:409-414. [ Links ]
10. Fathi M, Lundberg IE. Interstitial lung disease in polymyositis and dermatomyositis. Curr Opin Rheumatol 2005;17:701-706. [ Links ]
11. Leslie KO, Trahan S, Gruden J. Pulmonary Pathology of the Rheumatic Diseases. Seminars Respirat Crit Care Med 2007;28:369-378. [ Links ]
12. Targoff IN. Autoantibodies and Their Significance in Myositis. Current Rheumatology Reports 2008;10: 333-340. [ Links ]
13. Wells AU. Pulmonary Function Tests in Connective Tissue Disease. Semin Respir Crit Care Med 2007;28:379-388. [ Links ]
14. Greenberg SA. Inflammatory Myopathies: Evaluation and Management. Seminars Neurol 2008;28:241-249. [ Links ]
15. Devaraj A, Wells AU, Hansell DM. Computed Tomographic Imaging in Connective Tissue Diseases. Semin Respir Crit Care Med 2007;28:389-397. [ Links ]
16. Lakhanpal S, Lie JT, Conn DL, Martin II WJ. Pulmonary disease in polymyositis/dermatomyositis: a clinicopathological analysis of 65 autopsy cases. Ann Rheum Dis 1987;46:23-29. [ Links ]
17. Dubowitz LMS, Dubowitz V. Acute dermatomyositis presenting with pulmonary manifestations. Arch Dis Child 1964;39:293-296. [ Links ]
18. Aoun NY, Velez E, Aggarwal A, Hayes GB, Kenney LA. Fatal Acute Interstitial Pneumonitis Complicating Polymyositis in a 41-Year-Old Man. Respir Care 2004;49:1515-1521. [ Links ]
19. Cottin V, Thivolet-Béjui F, Reynaud-Gaubertz M, et al. Interstitial lung disease in amyopathic dermatomyositis, dermatomyositis and polymyositis. Eur Respir J 2003;22:245-250. [ Links ]
20. Marie I, Hachulla E, Hatron P-Y, et al. Polymyositis and Dermatomyositis: Short Term and Longterm Outcome, and Predictive Factors of Prognosis. J Rheumatol 2001;28:2230-2237. [ Links ]
21. Le Goff B, Che' Rin P, Cantagrel A, et al. Pneumomediastinum in Interstitial Lung Disease Associated With Dermatomyositis and Polymyositis. Arthritis & Rheumatism (Arthritis Care & Research) 2009;61:108-118. [ Links ]
22. Selva-O'Callaghan A, Labrador-Horrillo M, Muñoz-Gall X, et al. Polymyositis/dermatomyositis-associated lung disease: analysis of a series of 81 patients. Lupus 2005;14:534-542. [ Links ]

