










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Colombiana de Reumatología
versión impresa ISSN 0121-8123
Rev.Colomb.Reumatol. v.16 n.4 Bogotá oct./dic. 2009
PRESENTACIÓN DE CASO Y REVISIÓN DE LA LITERATURA
Bullous Systemic Lupus Erythematosus: dramatic response to dapsone
Luis Alonso González1, Mauricio Restrepo1, Gloria Vásquez1,2
1 Profesores, Sección de Reumatología, Departamento de Medicina Interna, Facultad de Medicina, Universidad de Antioquia, Hospital Universitario San Vicente de Paúl, Medellín, Colombia.
2 Grupos de Inmunología celular e inmunogenética. Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia.
E-mail: luisalonsogonzalez@une.net.com lagnvvn@hotmail.com
Recibido: Noviembre 15 de 2009 Aprobado: Diciembre 15 de 2009
Resumen
El lupus ampolloso es una manifestación poco frecuente del lupus eritematoso sistémico (LES). Otras enfermedades ampollosas tales como el penfigoide ampolloso, epidermólisis ampollosa adquirida, dermatosis ampollosa Ig A lineal y dermatitis herpetiforme también han sido informadas en LES. Describimos un paciente que desarrolló lesiones ampollosas 14 días luego de iniciar terapia con altas dosis de glucocorticoides y ciclofosfamida para manifestaciones severas del LES (nefritis y hemorragia alveolar). Se confirmó el diagnóstico de lupus ampolloso. La respuesta al tratamiento con dapsona fue notable a las 48 horas. En este artículo revisamos la epidemiología, hallazgos clínicos, histopatológicos e inmunopatológicos; el diagnóstico diferencial y el tratamiento del LES ampolloso.
Palabras clave: lupus eritematoso sistémico ampolloso; dapsona, lupus cutáneo.
Summary
Bullous systemic lupus erythematosus (BSLE) is an unusual finding in systemic lupus erythematosus (SLE). Other bullous disorders such as bullous pemphigoid, epirdermolysis bullosa acquisita, linear IgA bullous dermatosis and dermatitis herpetiformis has also been reported in association with SLE. We described a patient who developed severe bullous lesions 14 days after high-dose systemic glucocorticoids and cyclophosphide therapies were initiated for severe SLE manifestations (nephritis and alveolar hemorrhage). A diagnosis of bullous SLE was made. Therapy with dapsone resulted in a marked clinical improvement of the bullous eruption within 48 hours. This article also discusses the epidemiology, clinical, histopathologic and immunopathologic features, differential diagnosis and the treatment of BSLE.
Key words: bullous systemic lupus erythematosus; dapsone; cutaneous lupus.
Introducción
El compromiso cutáneo en los pacientes con lupus eritematoso sistémico (LES) es común y heterogéneo, presentándose hasta en el 90% de los pacientes1 y es la primera manifestación de la enfermedad en el 23% a 28% de los pacientes2. Según la clasificación de Gilliam3, las lesiones cutáneas del LES son específicas [LE cutáneo agudo, LE cutáneo subagudo (anular, papuloescamoso) y LE cutáneo crónico (LE discoide, LE hipertrófico o verrucoso, lupus profundus, LE tumidus y lupus sabañón o perniótico)] e inespecíficas. El lupus eritematoso sistémico ampolloso (LESA) hace parte de las manifestaciones cutáneas inespecíficas y es una rara enfermedad ampollosa subepidérmica mediada por anticuerpos4. Esta enfermedad se presenta como un brote vesículo-ampolloso diseminado, no cicatrizante que responde dramáticamente a dapsona; además, con una histología similar a la de la dermatitis herpetiforme y con características inmunológicas muy parecidas a las de la epidermólisis ampollosa adquirida (epidermolysis bullosa acquisita, EBA)5.
En el presente artículo, presentamos un paciente que desarrolla LESA luego de iniciar inmunosupresión con pulsos intravenosos de ciclofosfamida y de metilprednisolona para manifestaciones severas del LES (nefritis y hemorragia alveolar) y quien tiene una rápida respuesta al tratamiento con dapsona. Además, realizamos una revisión de la literatura sobre LESA.
Presentación del caso
Hombre de 19 años, quien es hospitalizado por cuadro clínico de mes y medio de evolución consistente en inflamación de rodilla derecha, edema en hemicara izquierda, adenomegalias cervicales, fiebre, pérdida no cuantificada de peso, úlceras orales, tos con escasa expectoración hemoptoica y disnea. Sin antecedentes personales o familiares importantes. Al examen físico: FC de 110', presión arterial 110/70mm Hg, y frecuencia respiratoria de 18'; además presentaba eritema malar, úlceras en paladar y sinovitis en rodillas; el resto del examen cardiopulmonar, abdominal, vascular y neurológico incluyendo fondo de ojo fue normal.
Las pruebas de laboratorio revelaron anemia (hemoglobina: 7.5 g/dl) normocítica normocrómica, linfopenia, hipocomplementemia (C3 27 mg/dl, C4 1.8 mg/dl), creatinina 0.7 mg/dl, proteinuria en 24 horas 2.8 gr, sedimento urinario activo (cilindros eritrocitarios, = 5 eritrocitos por campo de alto poder), derrame pericárdico, ANA (+) 1:640, patrón moteado, anti DNA de cadena doble (+) 1:320, anti Sm y RNP (+). Por caída inexplicada de la hemoglobina y aspecto de vidrio esmerilado difuso en una tomografía de tórax de alta resolución, se solicitó fibrobroncoscopia, la cual demostró sangrado activo escaso y hemosiderófagos en lavado broncoalveolar, confirmando una hemorragia alveolar. Se inició manejo con pulso intravenoso de ciclofosfamida (1 gramo), pulsos de metilprednisolona (500 mg/ día por 3 días), seguido por prednisolona oral (1 mg/kg/día), cloroquina, enalapril (10 mg/día), calcio y calcitriol. La histopatología de la biopsia renal confirma el diagnóstico de nefritis lúpica clase III (índice de actividad de 3/24 e índice de cronicidad de 0/12).
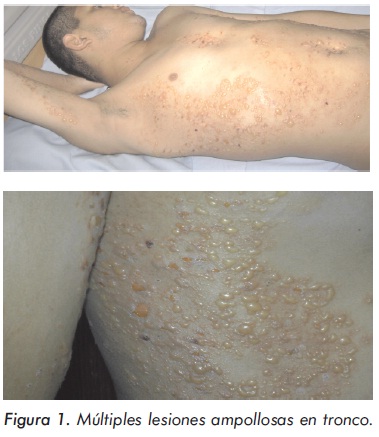
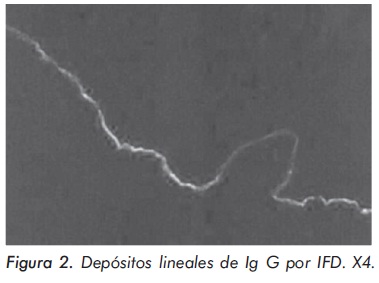
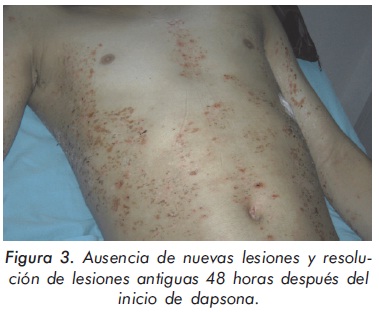
La respuesta al tratamiento fue satisfactoria y con ausencia de efectos adversos inmediatos. Catorce días después de recibir los pulsos de ciclofosfamida y metilprednisolona, aparecen vesículas y ampollas en tórax, abdomen, espalda y extremidades superiores (Figura 1). Ante la sospecha de infección diseminada por virus Varicela zoster, se inició aciclovir endovenoso. El informe de la biopsia de piel confirmó el diagnóstico de lupus ampolloso y en la inmunofluorescencia directa se observaron depósitos lineales de Ig G por IFD en la membrana basal (Figura 2) por lo cual se suspendió aciclovir y se inició dapsona 100 mg/día, presentando una dramática respuesta sin aparición de nuevas lesiones y rápida resolución de las previas después de 48 horas (Figura 3).
Dos meses después, durante el seguimiento ambulatorio, el paciente no presentaba evidencia de actividad clínica y serológica. Sin embargo, luego de suspender por una semana el tratamiento con dapsona, presentó de nuevo vesículas y ampollas en piel, que resolvieron luego de reiniciar tratamiento con dapsona (100 mg/día). El paciente persistió en remisión clínica de sus lesiones cutáneas bajo este tratamiento; además del uso de pulsos mensuales de ciclofosfamida indicados para el manejo de la nefritis lúpica clase III y la hemorragia alveolar.
Lupus Eritematoso Sistémico Ampolloso
Los pacientes con LES activo son propensos a desarrollar una enfermedad ampollosa autoinmune subepidérmica adquirida la cual no se clasifica como una de las dermatosis ampollosas autoinmunes primarias5. El brote cutáneo se caracteriza por ampollas que se originan en una base eritematosa o urticariforme semejando un penfigoide ampolloso o se puede presentar como vesículas agrupadas imitando una dermatitis herpetiforme6,7. El LESA es reconocido como una entidad separada asociada a autoinmunidad contra el colágeno tipo VII, un componente importante de las fibrillas de anclaje y antígeno de la EBA8,9. Los criterios inmunopatológicos propuestos por Yell y cols. del LESA son similares a los de la EBA. Por ejemplo, los depósitos de inmunoglobulinas y complemento se localizan en la membrana basal en la inmunofluorescencia directa (IFD) o en la inmunofluorescencia indirecta (IFI) y ultra-estructuralmente en o por debajo de la lámina densa10. Al igual que en la EBA, algunos de los pacientes con LESA tienen autoanticuerpos circulantes dirigidos contra el colágeno tipo VII5,9; sin embargo, a pesar de tal similitud inmunológica entre ambas entidades, existen algunas diferencias: (1) el LESA afecta principalmente a pacientes jóvenes, mientras que la EBA es más frecuente en la cuarta y quinta década de la vida; (2) las lesiones del LESA usualmente no dejan cicatrices, mientras que las de la EBA dejan cicatriz; (3) el LESA responde dramáticamente a la dapsona mientras que la EBA no5,11.
Algunos autores, han señalado que el colágeno tipo VII no es el único antígeno blanco en el LESA, sino que otros componente esenciales para la unión dermo-epidérmica (antígeno 1 del penfigoide ampolloso, laminina-5, laminina-6) también son un blanco antigénico.12 Además, algunos autores consideran que el LESA es una entidad heterogénea, que comprende todas la enfermedades ampollosas autoinmunes en las cuales hay una respuesta inmune dirigida contra elementos de la membrana basal13.
Criterios diagnósticos para LESA
Los primeros criterios para el diagnóstico de LESA fueron propuestos por Camisa y Sharma en 198314, (Tabla 1) y después revisados aplicando técnicas de inmunofluorescencia de la piel de la lesión15,16. Posteriormente, Yell y cols.10 revisaron estos criterios debido a la heterogeneidad de la presentación clínica e inmunohistológica de esta entidad y definieron el LESA como una enfermedad ampollosa en pacientes con LES, en la que los reactantes inmunes se encuentran presentes en la zona de la membrana basal en la IFD o en la IFI.

Epidemiología
El LESA es una enfermedad rara y su incidencia se ha estimado en 0.2 casos por millón de habitantes, de acuerdo con un estudio francés17. Entre 324 pacientes con enfermedades ampollosas inmunológicas adquiridas, diagnosticadas en un período de 15 años, el 1.5% tuvieron LESA18. El LESA afecta especialmente a adultos jóvenes entre la segunda y cuarta década de la vida, aunque también se ha informado casos en adultos mayores19. Las mujeres, particularmente de raza negra, son más afectadas que los hombres. También puede presentarse en cualquier grupo étnico. El predominio en mujeres jóvenes posiblemente sólo refleje el patrón de distribución habitual del LES en la población20.
Hallazgos clínicos
El LESA se caracteriza por el inicio agudo de un brote ampolloso generalizado, pruriginoso, que por lo general no deja cicatriz. El brote puede aparecer en cualquier sitio de la piel; sin embargo, el tronco superior, el cuello, las regiones supraclaviculares, los pliegues axilares, la parte proximal de las extremidades (tanto superficies flexoras como extensoras) son las áreas de predilección. Las áreas expuestas al sol son las más afectadas, aunque las lesiones también se pueden presentar en zonas no expuestas al sol. Las lesiones incluyen ampollas, vesículas y un brote maculopapular. Las ampollas pueden surgir sobre una piel eritematosa o normal; son tensas, con líquido claro o hemorrágico y ocasionalmente se rompen dejando erosiones, costras y cambios pigmentarios (máculas hipo o hiperpigmentadas). Por lo general estas lesiones son múltiples, se expanden rápidamente hacia la periferia y se unen formando figuras alargadas e irregulares (Figura 1). Dependiendo del predominio de las lesiones inflamatorias y la distribución de la erupción, esta última puede imitar un penfigoide ampolloso (PA), dermatitis herpetiforme (DH) o la variante inflamatoria de la EBA5,13,18.
En el LESA, la presencia de fragilidad cutánea, ampollas traumáticas, cicatrices y milia características de la variante clásica de la EBA por lo general están ausentes; sin embargo, más de una decena de casos informados de LESA se han presentado con hallazgos de EBA12,21-26.
Usualmente, los pacientes con LESA, presentan actividad lúpica en otros órganos7,27,28, especialmente nefritis lúpica16,28,29; sin embargo, el inicio y la evolución de las lesiones cutáneas pueden presentarse en ausencia de actividad lúpica en otros sistemas5. En algunos casos, el brote cutáneo aparece entre 4 y 12 días luego del inicio de glucocorticoides sistémicos30,31, como sucedió en nuestro paciente.
Diagnóstico diferencial
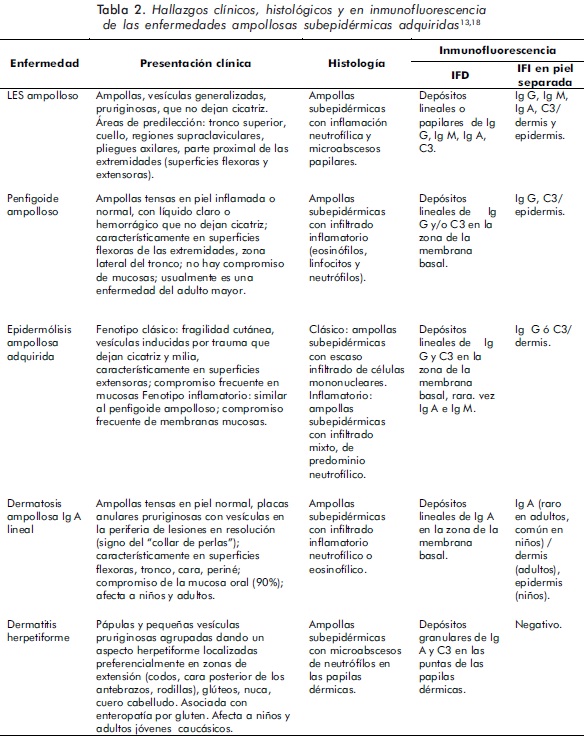
Algunas enfermedades ampollosas subepidérmicas adquiridas, tales como el PA, la EBA, la DH y la dermatosis ampollosa Ig A lineal, han sido informadas en pacientes con LES y pueden ser confundidas con un LESA. Su diferenciación se basa en los hallazgos clínicos, histológicos e inmunopatológicos (Tabla 2)13,18.
Histopatología
Los hallazgos característicos son las ampollas subepidérmicas con microabscesos de neutrófilos en las papilas dérmicas, hallazgos similares a los de la DH5,6,11,13,14. La cavidad de las ampollas contiene fibrina y una gran cantidad de neutrófilos18. La dermis se encuentra edematosa. Los neutrófilos pueden estar distribuidos uniformemente en un patrón en banda dentro de la dermis papilar. Un infiltrado inflamatorio perivascular moderado envuelve los vasos de la dermis superficial y media, el cual consiste principalmente de linfocitos, aunque también puede contener neutrófilos y eosinófilos. En ocasiones se observa vasculitis leucocitoclástica y extravasación de eritrocitos13. Otro hallazgo histológico que permite distinguir el lupus ampolloso de otras enfermedades ampollosas inmunológicas es la presencia de mucina en la dermis reticular31.
Inmunopatología
La característica inmunopatológica del LESA es el depósito de reactantes inmunes a lo largo de la unión dermoepidérmica demostrado mediante IFD sobre piel perilesional y sobre piel clínicamente no afectada18. La tinción por inmunofluorescencia también se puede detectar en la dermis superior y ocasionalmente en los vasos de la dermis superior. Los depósitos inmunes contienen Ig G, Ig M, e Ig A; sin embargo, la Ig G está presente en todos los casos13,18. La Ig M se encuentra en la mitad de los pacientes13. Los depósitos de Ig A son más frecuentes en el lupus ampolloso que en lupus no ampolloso (76% vs 17%)32. Los componentes del complemento frecuentemente se detectan en biopsias de lesiones cutáneas y rara vez en la piel sin compromiso clínico33.
El depósito de reactantes inmunes en la zona de la membrana basal tiene dos patrones: un patrón granular, presente en el 40% de los casos, y un patrón lineal, comparable con la banda lúpica en el 60% restante13. La IFD puede ser útil para descartar DH en la cual es más característico el depósito granular aislado de IgA debajo de la membrana basal. Si los depósitos de IgA son lineales y homogéneos se debe considerar una dermatosis ampollosa IgA lineal. La presencia concomitante de IgG e IgM está más a favor de LESA. Sin embargo, la IFD y la IFI no distinguen entre LESA y PA en piel intacta. Utilizando como sustrato piel separada en NaCl 1 M, en la IFI se pueden observar los depósitos de anticuerpos en el techo de la ampolla en el PA34, mientras que en el LESA estos se observan en el piso o lado dérmico de la ampolla8,35. En estudios ultraestructurales el depósito de reactantes inmunes se localiza en o por debajo de la lámina densa, similar a lo observado en EBA8,11.
En pacientes con hallazgos clínicos, histológicos y de inmunofluorescencia de LESA, la IFI en piel normal muestra resultados contradictorios respecto a la presencia de anticuerpos circulantes anti-membrana basal, lo cual ha llevado a establecer dos subtipos de LESA inmunológicamente diferentes, el LESA tipo I y el LESA tipo II, los cuales se caracterizan por la presencia o ausencia de anticuerpos circulantes y/o unidos a tejido dirigidos contra el colágeno tipo VII de la membrana basal, respectivamente5.
Análisis de inmunoblot y patogénesis
Mediante el análisis de inmunoblot, anticuerpos Ig G anti membrana basal se unen a dos antígenos de diferente peso molecular, las proteínas de 290 y 145 kDa de la cadena alfa del colágeno tipo VII, tanto en la dermis normal como en el suero de los pacientes con LESA18,36,37.
El colágeno tipo VII es el principal componente de las fibrillas de anclaje de la unión dermoepidérmica. Los epítopes antigénicos principales para los autoanticuerpos en pacientes con lupus ampolloso se han demostrado dentro del dominio no colágeno aminoterminal NC1 del colágeno tipo VII, justamente dentro de su región de homología a la fibronectina tipo III, la cual media la interacción entre las fibrillas de anclaje y otras proteínas de matriz38,39. Además de reaccionar con el NC1, el suero de pacientes con LESA y EBA también reacciona con el dominio no colágeno carboxiterminal NC2 del colágeno tipo VII, cuando se analiza utilizando las técnicas de ELISA e Inmunoblot utilizando NC2 recombinante como antígeno.
Se han propuesto varios mecanismos para explicar la formación de ampollas a través de estos autoanticuerpos contra colágeno tipo VII que incluyen: (1) interferencia con las interacciones normales entre el colágeno tipo VII y sus ligandos de la matriz extracelular en la membrana basal o en la dermis papilar, lo cual debilita o bloquea las conexiones de las fibrillas de anclaje con la lámina densa o las placas de anclaje, lo cual finalmente lleva a una adhesión defectuosa dermis- lámina densa40; (2) los anticuerpos unidos al dominio NC2 desestabilizan las fibrillas de anclaje al interferir con la formación de dímeros antiparalelos del colágeno tipo VII41; (3) un tercer mecanismo es el daño tisular inflamatorio por activación del complemento42.
También se han informado la presencia de otros autoanticuerpos que reaccionan con otros antígenos en la región de la membrana basal tales como el antígeno 1 del penfigoide ampolloso, lamimina 5 y lamimina 612. Esto puede explicarse por el fenómeno inmunológico de diseminación del epítope. Este fenómeno describe un evento inmunológico en el cual un proceso autoinmune o inflamatorio primario produce una injuria tisular, que lleva a la liberación de epítopes antigénicos normalmente ocultos contra los cuales se genera una respuesta autoinmune secundaria. Por lo tanto, la reacción autoinmune primaria contra el colágeno tipo VII puede inducir una reacción inmune secundaria contra laminina 5 y otros componente de membrana basal12. La presencia de anticuerpos contra varios componentes de la zona de la membrana basal como consecuencia de la diseminación del epítope, puede ser la explicación para la heterogeneidad en el fenotipo clínico y el perfil inmunológico del LESA18.
La predisposición genética también puede ser responsable de un alto riesgo de desarrollar una respuesta autoinmune contra los antígenos de la zona de la membrana basal. Comparado con la población normal, tanto los pacientes con LESA como con EBA, tienen una alta incidencia del haplotipo HLA-DR243.
Tratamiento
Una característica clínica importante que diferencia el LESA de la EBA es su notable respuesta terapéutica a la dapsona5,11,44,45. Generalmente, la mejoría es dramática con el cese de la formación de nuevas ampollas en 24 a 48 horas luego de iniciar el tratamiento y resolución completa de las lesiones a los 7 a 10 días6,11,46. Dosis bajas (25-50 mg/día) son usualmente efectivas, aunque a veces se requieren dosis más altas (100 mg/día)16,18. Rápidas recaídas pueden presentarse luego de suspender el tratamiento con dapsona, sin embargo las lesiones desaparecen rápidamente luego de reiniciar la dapsona. La suspensión del tratamiento con dapsona por lo general es posible un año luego de haberlo iniciado. A diferencia de la DH, la disminución gradual y posterior suspensión de dapsona no siempre resulta en recaída de la enfermedad11,37. Los glucocorticoides sistémicos a altas dosis y los inmunosupresores utilizados para el compromiso sistémico, usualmente son inefectivos para el manejo del LESA47. En algunos casos que no toleran o responden a dapsona, se han utilizado glucocorticoides en dosis altas y azatioprina14,16,48,49. También se ha utilizado ciclofosfamida16, sulfapiridina50 y metotrexate28 pero la experiencia es limitada.
Referencias
1. Petri M. Dermatologic lupus. Sem Cut Med Surg. 1998;17:219-227. [ Links ]
2. Pistiner M, Wallace DJ, Nessim S, et al. Lupus erythematosus in the 1980s: a survey of 570 patients. Semin Arthritis Rheum. 1991;21:55-64. [ Links ]
3. Gilliam JN, Sontheimer RD. Skin manifestations of SLE. Clin Rheum Dis. 1982;8:207-218. [ Links ]
4. Costner MI, Sontheimer RD. Lupus-Nonspecific Skin Disease. In: Wallace DJ, Hahn BH, eds. Dubois' Lupus Erythematosus. 7th ed. Los Angeles, California: Lippincott Williams & Wilkins; 2007:621-636. [ Links ]
5. Gammon WR, Briggaman RA. Bullous SLE: a phenotypically distinctive but immunologically heterogeneous bullous disorder. J Invest Dermatol. 1993;100:28S-34S. [ Links ]
6. Burrows NP, Bhogal BS, Black MM, et al. Bullous eruption of systemic lupus erythematosus: a clinicopathological study of four cases. Br J Dermatol. 1993;128:332-338. [ Links ]
7. Rappersberger K, Tschachler E, Tani M, Wolff K. Bullous disease in systemic lupus erythematosus. J Am Acad Dermatol.1989;21:745-752. [ Links ]
8. Gammon WR, Woodley DT, Dole KC, et al. Evidence that anti-basement membrane zone antibodies in bullous eruption of systemic lupus erythematosus recognize epidermolysis bullosa acquisita autoantigen. J Invest Dermatol. 1985;84:472-476. [ Links ]
9. Chen M, Chan LS, Cai X, et al. Development of an ELISA for rapid detection of anti-type VII collagen autoantibodies in epidermolysis bullosa acquisita. J Invest Dermatol 1997;108:68-72. [ Links ]
10. Yell JA, Allen J, Wojnarowska F, et al. Bullous systemic lupus erythematosus: revised criteria for diagnosis. Br J Dermatol 1995;132:921-928. [ Links ]
11. Hall RP, Lawley TJ, Smith HR, Katz SI. Bullous eruption of systemic lupus erythematosus. Dramatic response to dapsone therapy. Ann Intern Med 1982;197:165-170. [ Links ]
12. Chan LS, Lapiere JC, Chen M, et al. Bullous systemic lupus erythematosus with autoantibodies recognizing multiple skin basement membrane components, bullous pemphigoid antigen 1, laminin-5, laminin- 6, and type VII collagen. Arch Dermatol 1999; 135:569-573. [ Links ]
13. Yell JA, Wojnarowska F. Bullous skin disease in lupus erythematosus. Lupus 1997;6:112-121. [ Links ]
14. Camisa C, Sharma HM. Vesiculobullous systemic lupus erythematosus. Report of two cases and a review of the literature. J Am Acad Dermatol. 1983; 9:924- 933. [ Links ]
15. Camisa C, Grimwood RE. Indirect immunofluorescence in vesiculobullous eruption of systemic lupus erythematosus. J Invest Dermatol. 1986; 86:606. [ Links ]
16. Camisa C. Vesiculobullous systemic lupus erythematosus. A report of four cases. J Am Acad Dermatol. 1988;18:93-100. [ Links ]
17. Bernard P, Vaillant L, Labeille B, et al. Incidence and distribution of subepidermal autoimmune bullous skin diseases in three French regions. Arch Dermatol 1995;131:48-52. [ Links ]
18. Vassileva S. Bullous systemic lupus erythematosus. Clin Dermatol. 2004;22:129-138. [ Links ]
19. Wong SN, Chua SH. Spectrum of subepidermal immunobullous disorders seen at the National Skin Centre, Singapore: a 2-year study. Br J Dermatol 2002;147:476-480. [ Links ]
20. Jablonska S, Blaszczyk M. Connective tissue diseases. In: Parish LCP, Brenner S, Ramos-e-Silva M, eds. Women's dermatology: from infancy to maturity. New York, London:The Partenon Publishing Group, 2001:205-217. [ Links ]
21. Dotson AD, Raimer SS, Pursley TV, et al. Systemic lupus erythematosus occurring in a patient with epidermolysis bullosa acquisita. Arch Dermatol 1981;117:422-426. [ Links ]
22. McHenry PM, Dagg JH, Tidman JH, et al. Epidermolysis bullosa acquisita occurring in association with bullous lupus erythematosus. Clin Exp Dermatol 1991;1993:378-380. [ Links ]
23. Boh E, Roberts LJ, Lieu TS, et al. Epidermolysis bullosa acquisita preceding the development of systemic lupus erythematosus. J Am Acad Dermatol 1990;22:587- 593. [ Links ]
24. Don PC. Vesiculobullous lupus erythematosus with milia formation. Int J Dermatol 1992;31:793-795. [ Links ]
25. Prussick R, Gupta AK. Epidermolysis bullosa acquisita with features of bullous lupus erythematosus. Int J Dermatol 1994;33:192-195. [ Links ]
26. Eckman JA, Mutasim DF. Bullous systemic lupus erythematosus with milia and calcinosis. Cutis 2002;70:31-34. [ Links ]
27. Miyagawa S, Shiomi Y, Fukumoto T, et al. Bullous eruption of systemic lupus erythematosus. J Dermatol 1994;21:421-425. [ Links ]
28. Malcangi G, Brandozzi G, Giangiacomi M, Zampetti M, Danieli Mg. Bullous SLE: response to methotrexate and relationship with disease activity. Lupus 2003;12:63-66. [ Links ]
29. Ng YY, Chang T, Chen TW, Liou HN, Yang AH, Yang WC. Concomitant lupus nephritis and bulous eruption in systemic lupus erythematosus. Nephrol Dial Transplant 1999;14:1739-1743. [ Links ]
30. Lalova A, Pramatarov K, Vassileva S. Facial bullous systemic lupus erythematosus. Int J Dermatol 1997;36:356-373. [ Links ]
31. Tsuchida T, Furue M, Kashiwado T, et al. Bullous systemic lupus erythematosus with cutaneous mucinosis and leukocytoclastic vasculitis. J Am Acad Dermatol 1994;31:387-390. [ Links ]
32. Crowson AN, Magro C. The cutaneous pathology of lupus erythematosus: a review. J Cutan Pathol. 2001;28:1-23. [ Links ]
33. Vassileva S. Lupus erythematosus. In: Kanitakis J, Vassileva S, Woodley D, eds. Diagnostic immunohistochemistry of the skin. London: Chapman & Hall, 1998:144-156. [ Links ]
34. Woodley D, Sauder D, Talley MJ, Silver M, Grotendorst G, Qwarnstrom E. Localization of basement membrane components after dermal-epidermal junction separation. J Invest Dermatol. 1983;81:149- 153. [ Links ]
35. Janniger CK, Kowalewski C, Mahmood T, Lambert WC, Schwartz RA. Detection of anti-basement membrane zone antibodies in bullous systemic lupus erythematosus. J Am Acad Dermatol. 1991;24:643- 647. [ Links ]
36. Barton DD, Fine JD, Gammon WR, Sams WM Jr.. Bullous systemic lupus erythematosus: an unusual clinical course and detectable circulating autoantibodies to the epidermolysis bullosa acqusita antigen. J Am Acad Dermatol 1986;15:369-373 [ Links ]
37. Shirahama S, Furukawa F, Yagi H, Tanaka T, Hashimoto T, Takigawa M. Bullous systemic lupus erythematosus: detection of antibodies against noncollagenous domain of type VII collagen. J Am Acad Dermatol 1998;38:844-848. [ Links ]
38. Jones DA, Hunt SW 3rd, Prisayanh PS, Briggaman RA, Gammon WR. Immunodominant autoepitopes of type VII collagen are short, paired peptide sequences within the fibronectin type III homology region of the noncollagenous (NC1) domain. J Invest Dermatol 1995;104:231-235. [ Links ]
39. Chen M, Marinkovich MP, Jones JC, O'Toole EA, Li YY, Woodley DT.NC1 domain of type VII collagen binds to the beta3 chain of laminin 5 via a unique subdomain within the fibronectin-like repeats. J Invest Dermatol. 1999;112:177-183. [ Links ]
40. Gammon WR. Autoimmunity to collagen VII: autoantibody- mediated pathomechanisms regulate clinical-pathological phenotypes of acquired epidermolysis bullosa and bullous SLE. J Cutan Pathol 1993;20:109-114. [ Links ]
41. Chen M, Keene DR, Costa FK, et al. The carboxyl terminus of type VII collagen mediates antiparallel dimer formation and constitutes a new antigenic epitope for epidermolysis bullosa acquisita autoantibodies. J Biol Chem 2001;276:21649- 21655. [ Links ]
42. Gammon WR, Briggaman RA, Inman AO III, Merritt CC, Wheeler CE Jr. Evidence supporting a role for immune complex-mediated inflammation in the pathogenesis of bullous lesions of systemic lupus erythematosus. J Invest Dermatol 1983;81:320-325. [ Links ]
43. Gammon WR, Heise ER, Burke WA, Fine JD, Woodley DT, Briggaman RA. Increased frequency of HLA-DR2 in patients with autoantibodies to epidermolysis bullosa acquisita antigen. Evidence that the expression of autoimmunity to type VII collagen is HLA class II allele associated. J Invest Dermatol 1988;91:228-232. [ Links ]
44. Lance NJ, Blaszak W, Swartz TJ. Bullous skin lesions in systemic lupus erythematosus. Sem Arthritis Rheum 1991;20:396-404. [ Links ]
45. Tani M, Shimizu R, Ban M, Murata Y, Tamaki A. Systemic lupus erythematosus with vesiculobullous lesions. Arch Dermatol 1984;120:1497-1501. [ Links ]
46. Yung A. Oakley A. Bullous systemic lupus erythematosus. Australas J Dermatol 2000;41:234-237. [ Links ]
47. Patricio P, Ferreira C, Gomes MM, Filipe P. Autoimmune Bullous Dermatoses: A Review. Ann NY Acad Sci 2009;1173:203-210. [ Links ]
48. Olansky AJ, Briggaman RA, Gammon WR, Kelly TF, Sams WM Jr. Bullous systemic lupus erythematosus. J Am Acad Dermatol. 1982;7:511-520. [ Links ]
49. Jacoby RA, Abraham AA. Bullous dermatosis and systemic lupus erythematosus in a 15-year-old boy. Arch Dermatol. 1979;115:1094-1097. [ Links ]
50. Kettler AH, Bean SF, Duffy JO, Gammon WR. Systemic lupus erythematosus presenting as a bullous eruption in a child. Arch Dermatol. 1988;124:1083-1087. [ Links ]

