










Servicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Colombiana de Reumatología
versión impresa ISSN 0121-8123
Rev.Colomb.Reumatol. v.17 n.2 Bogotá abr./jun. 2010
ARTÍCULO DE REVISIÓN
Autoinflammatory Diseases
Édgar Peñaranda-Parada1, Néstor Spinel-Bejarano2, José Félix Restrepo3, Federico Rondón-Herrera4, Alberto Millán5 Antonio Iglesias Gamarra3
1 MD. Residente Reumatología, Universidad Nacional de Colombia.
2 MD. Residente Reumatología, Universidad Nacional de Colombia.
3 MD. Especialista en Reumatología. Profesor titular Facultad de Medicina, Universidad Nacional.
4 MD. Especialista en Reumatología. Profesor asociado Facultad de Medicina, Universidad Nacional.
5 Internista-Reumatólogo. Venezuela.
Recibido: Abril 05 de 2010 Aceptado: Mayo 18 de 2010
Resumen
Presentamos un artículo de revisión sobre las enfermedades autoinflamatorias, narrando su origen histórico y describiendo la estructura proteica y molecular del Inflamosoma, la clasificación actual de los trastornos autoinflamatorios y una descripción de las características inmunogenéticas y clínicas más sobresalientes de cada enfermedad.
Palabras clave: enfermedades autoinflamatorias, inflamosoma, inflamosomopatías, criopirina.
Summary
We present a review article on the autoinflammatory diseases, narrating its historical origin and describing the protein and molecular structure of the Inflammasome, the current classification of the autoinflammatory diseases and a description of the immunogenetics and clinical characteristics more important of every disease.
Key words: autoinflammatory diseases, inflammasome, inflammasomopathies, cryopyrin.
Definición
Las enfermedades autoinflamatorias (EAI) se caracterizan por la presentación de brotes periódicos de inflamación sistémica que comprometen varios tejidos como articulaciones y sistemas como el gastrointestinal, el neurológico y la piel. A diferencia de las enfermedades autoinmunes (órgano específicas y no órgano específicas), no se asocian a una respuesta celular T con antígenos definidos, ni a altos títulos de anticuerpos; pero la agrupación de estas enfermedades probablemente está relacionada con el sistema inmune innato. Las manifestaciones febriles y reumáticas son comunes y pueden comprometer cualquier articulación o partes del sistema musculoesquelético. También, pueden simular patologías autoinmunes, infecciosas y neoplásicas.
El término criopirinopatía es usado comúnmente para designar a los desórdenes autoinflamatorios, pero en nuestro concepto no es apropiado porque estas enfermedades no sólo se asocian a mutaciones del gen de las pirinas, sino que existen múltiples alteraciones en diversos genes, asociadas a las enfermedades autoinflamatorias.
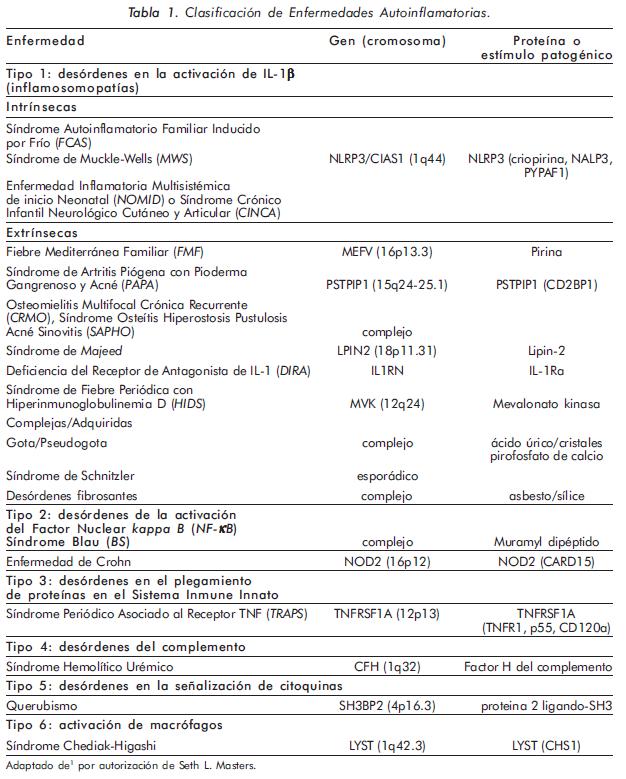
El concepto de enfermedad autoinflamatoria fue propuesto en 1999 por el equipo de Daniel Kastner (especializado en el estudio de enfermedades hereditarias caracterizadas por fiebres intermitentes); diez años después, el mismo Kastner, en vista de la diversidad de enfermedades similares que se estaban describiendo a nivel genético, planteó con expectación el concepto de "Horror autoinflamatorio", opinión muy similar a la planteada por Paul Ehrlich en 1897 sobre el "Horror autotóxico" para describir el inicio del concepto de autoinmunidad, el cual no fue entendido hasta más de medio siglo después1. Con el advenimiento y desarrollo de la biología molecular, en los últimos diez años se ha descrito una serie de patologías autoinflamatorias, que es nuestro deber detectar y diagnosticar. La nosología de las EAI ha avanzado rápidamente al reconocer enfermedades derivadas de variantes genéticas del sistema inmune innato. Los estudios moleculares de los mecanismos implicados en la génesis de estas patologías han desbordado los aspectos genéticos e inmunológicos, por lo que se ha planteado una clasificación de acuerdo a los mecanismos moleculares y funcionales1 (Tabla 1).
Origen
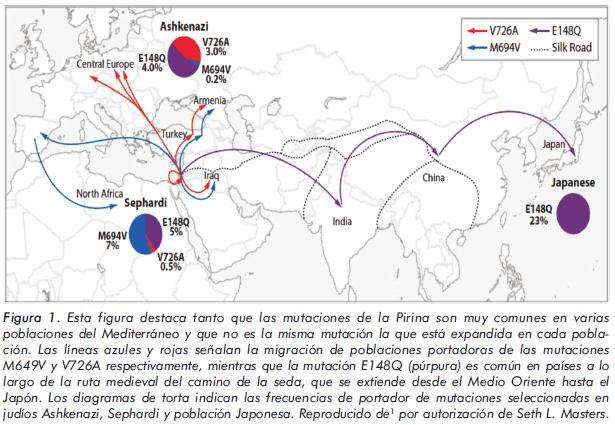
El origen de las EAI debe ser muy antiguo; posiblemente se remonta a los tiempos bíblicos, en los inicios de los pueblos de Israel, Palestina, Egipto, Turquía, Líbano, Siria y los países del área del Mediterráneo. La primera de estas enfermedades que se conoce es la Fiebre Mediterránea Familiar y el efecto del gen fundador a nivel de los genes V726A y M694V, originados en ancestros comunes que vivieron aproximadamente hace 2500 años en el área oriental del Mediterráneo (Figura 1). Posiblemente esta mutación originó un retraso en la aparición o reconocimiento de la enfermedad, porque esta solamente se empezó a describir en el año de 1908 por Janeway y Mosenthal, en una niña de origen judío que presentó dolor abdominal y fiebre. Los estudios se han centrado en las mutaciones del dominio de las Pirinas, que son comunes en poblaciones ancestrales del Mediterráneo y para ello se ha analizado el dominio de la Pirina B30.2/SPRY, el cual no se conserva en las especies inferiores, sugiriendo que estas mutaciones son de reciente aparición.
A través de la ontogenia y la filogenia del sistema inmunitario los vertebrados detectan los patógenos activando los sistemas innato y adaptativo; sin embargo, cuando el sistema inmune se sobreactiva, el sistema adaptativo se pone en alerta generando la presentación antigénica a través de los linfocitos B, T y macrófagos, produciéndose una serie de re-arreglos e hipermutación somáticas capaces de establecer un repertorio de receptores antigénicos que reconocen los presentes y futuros microorganismos. En contraste, el sistema inmunitario innato se caracteriza por su habilidad para reconocer una gama de patógenos como bacterias, virus y hongos a través de un número limitado de receptores codificados por la línea germinal denominada Receptores de Reconocimiento de Patrones (Pattern-recognition Receptors: PRRs). Estos receptores se expresan en células primitivas del sistema inmune innato como los neutrófilos y otras líneas más avanzadas como las del sistema monocito-macrófago, células dendríticas y epiteliales que permiten la detección temprana en el sitio de la infección. Estos PRRs reconocen estructuras de microorganismos a través de los patrones moleculares asociados a patógenos o PAMPs, que al activarse inician la producción de una serie de citoquinas y quemoquinas que generan la expresión de moléculas co-estimuladoras y de adhesión que atraen células del sistema inmunitario adaptativo al sitio de la infección y alertan a los patrones moleculares asociados a peligro (DAMPs) que se liberan en las células lesionadas. Durante la ontogenia del sistema inmunitario se van generando una serie de familias importantes como la superfamilia de inmunoglobulinas, selectinas, integrinas y caderinas, que juegan un papel fundamental en la regulación inmunitaria adaptativa y tienen una restricción genética importante. Durante la evolución también se van a desarrollar una serie de familias como los PAPMs, DAMPs, los NLR (Nodlike receptor) y los RLH (Rig-like helicasa). Los PAPMs y DAMPs son sensores de los diferentes microorganismos, urato monosódico, ATP, asbesto y cristales de pirofosfato, que al activarse generan el inflamosoma y el sistema NF-kB, que activados en forma inapropiada, en algunos casos por mutaciones genéticas, originan las diferentes enfermedades autoinflamatorias. Estas familias como las NLR, constituyen la familia Caterpiller2; las otras proteínas como las RLH, detectan virus y al parecer no se ha descubierto que generen alguna enfermedad de tipo autoinflamatorio. De los primeros estudios genéticos publicados en el 2001 por Hoffman, se identificó al gen responsable que denominó CIAS1 (coldinduced autoinflammatory syndrome 1), localizado en el cromosoma 1q44, que induce la síntesis de NALP3 o criopirinas3. Las mutaciones de genes ancestrales en el dominio NACHT, especialmente en el exón 3, constituyen algunos de los síndromes autoinflamatorios que fueron los primeros en describirse4.
El Inflamosoma
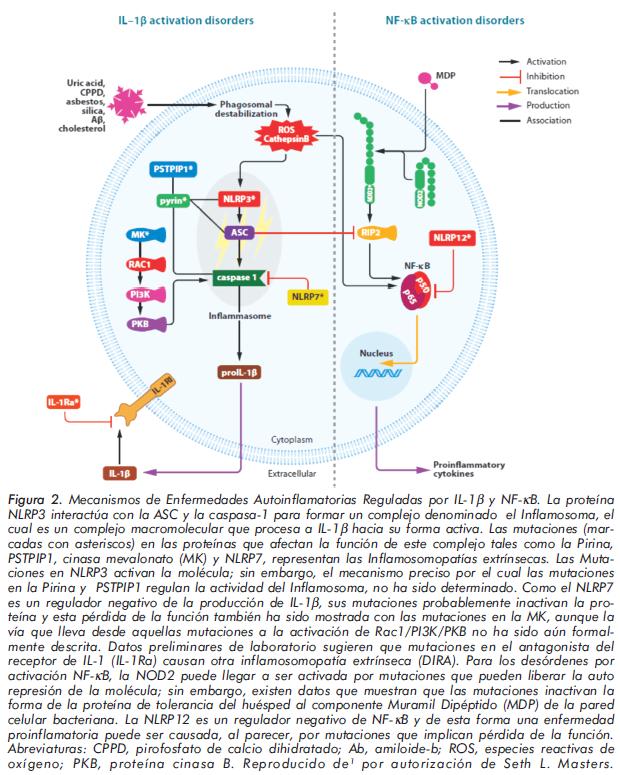
El Inflamosoma es un complejo de multi-proteínas que median la activación de la caspasa- 1, la cual promueve la secreción de las citocinas pro-inflamatorias interleucina-1β (IL-1β) e interleucina-18, así como la Piroptosis, una forma de muerte celular programada inducida por patógenos bacterianos. Miembros de la familia de receptores Nod-like (NLR), incluyendo el NLRP1, NLRP3, NLRC4 y el adaptador ASC, son componentes críticos del Inflamosoma, que acoplan estímulos bacterianos y endógenos a la activación de la caspasa-15.
Las caspasas son una familia de cisteínproteasas intracelulares que realizan clivaje a un limitado número de substratos a nivel de un residuo de ácido aspártico. Las caspasas son esenciales en la activación e implementación de la muerte celular y en el procesamiento y secreción de moléculas pro-inflamatorias. La caspasa-1, la primera en ser identificada, está presente en el citosol de células fagocíticas como un cimógeno inactivo, después de su estimulación por señales endógenas y bacterianas, el cimógeno procaspasa- 1 es autoactivado por clivaje proteolítico hacia un heterodímero enzimáticamente activo. La caspasa-1 es esencial para el clivaje de prointerleucina- 1β (pro-IL-1β) y pro-interleucina-18 (pro-IL-18) a sus formas maduras y biológicamente activas. Los miembros de la familia de las citocinas IL-1β difieren de otras citocinas en la falta de una secuencia génica líder y en que son expresadas como pro-formas biológicamente inactivas al citosol5,6. La IL-1β está asociada a varias reacciones inmunes, incluyendo el reclutamiento de células inflamatorias en el sitio de la infección mientras que la IL-18 es importante para la producción de interferón-γ y la amplificación de la actividad citolítica de las células asesinas naturales5,7. En la activación de la caspasa-1 es fundamental el ensamblaje de un gran complejo macromolecular a través de uniones de dominios reclutadores de caspasa (CARD), dominios Pirina e interacciones proteicas para formar el andamiaje del reclutamiento y la activación de la procaspasa-1. Esta plataforma molecular para la activación de la caspasa-1, la cual incluye miembros de la familia NLR y el adaptador ASC, ha sido llamada el Inflamosoma8. Las familias de proteínas NLR están comúnmente organizadas en 3 dominios: un dominio de repetición rico en leucina C-terminal (LRR), un dominio intermedio de oligomerización y unión a nucleótidos (NOD o NACHT) y otro conformado por una Pirina N-terminal (PYD) y un dominio reclutador y activador de caspasa (CARD). Los dominios LRR interactúan con ligandos putativos y juegan un papel en la auto-regulación de estas proteínas. Los dominios NOD o NACHT pueden ligar ribonucleótidos, los cuales regulan la autooligomerización y ensamblaje del Inflamosoma. Los dominios N-terminales median la interacción proteína-proteína con intermediarios de señalización y también son usados para subcategorizar las proteínas NLR6.
El Inflamosoma humano NLRP1 fue la primera plataforma molecular activada por caspasa-1 en ser identificada. El NLRP1 forma oligómeros con la caspasa-1 en la presencia de dipéptidos muramil (MDP) bacterianos. Varias bacterias Gram negativas, incluyendo Salmonella typhimurium, L. pneumophila, Pseudomonas aeruginosa y Shigella flexneri, inducen la activación de la caspasa-1 y la forma específica de muerte celular programada llamada Piroptosis por medio del Inflamosoma NLRC4, mecanismos activados por la llegada al citosol de la flagelina bacteriana. La activación de NLRC4 dependiente de la caspasa-1 está acompañada por la secreción de IL-1β y la inducción subsecuente de Piroptosis5,9.
NLRP3 y enfermedad: generalidades
El Inflamosoma NLRP3 (Criopirina) es activado por estímulos bacterianos, incluyendo Lipopolisacáridos, MDP y RNA. Una característica importante del NLRP3 es su activación por moléculas como: cristales de urato, ATP, asbesto, sílice y toxinas bacterianas formadoras de poros10,11. Los cristales de urato monosódico y de pirofosfato de calcio son activadores potenciales de la caspasa-1 a través del Inflamosoma NLRP3 y su activación por medio de los cristales de urato está implicada en la patogénesis de la inflamación por gota, lo cual es consistente con el hallazgo de mejoría clínica cuando los pacientes con ataques de gota son tratados con inhibidores del receptor de IL-111-13. Otros estímulos implicados en la activación del Inflamosoma NLRP3 son el hidróxido de aluminio y el amiloide fibrilar, una molécula asociada en la patogénesis de la enfermedad de Alzheimer.Dada la importancia de la IL-1β en los procesos que median la inflamación, no es inesperado que la desregulación de la activación del Inflamosoma esté relacionada con la patogénesis de una variedad de enfermedades inflamatorias. Las mutaciones en los genes que codifican los componentes del Inflamasoma, y de aquellos asociados al mismo, están implicadas en patologías como los síndromes febriles periódicos, Vitíligo y enfermedad de Crohn. Además, la producción de IL-1β dependiente del inflamosoma, está involucrada en la patogénesis de gota, pseudogota, asbestosis, silicosis y enfermedad de Alzheimer5. Las mutaciones autosómicas dominantes en NLRP3 están asociadas con un grupo de raros desórdenes autoinflamatorios, incluyendo el Síndrome Autoinflamatorio Familiar inducido por frío, el Síndrome de Muckle-Wells y el Desorden Inflamatorio Multisistémico de Inicio Neonatal/NOMID [llamados en conjunto Síndromes Periódicos Asociados a Criopirina/NLRP3 (CAPS) o Criopirinopatías]; los cuales comparten características clínicas dadas por episodios febriles recurrentes, rash y artropatías. Las enfermedades asociadas a mutaciones de NLRP3, resultan por la amplificación de la actividad de la caspasa-1 y secreción de IL-1β, causadas por activación constitutiva del Inflamosoma NLRP38. Los síndromes febriles periódicos hereditarios están genéticamente determinados y transmitidos acorde a las leyes mendelianas. Los ataques de fiebre e inflamación ocurren repetidamente, en ausencia de una causa detectable. Otros ejemplos incluyen: Síndrome Periódico Asociado al receptor de TNF (TRAPS), Síndrome Hiper Ig-D (HIDS) y la Fiebre Mediterránea Familiar (FMF) (Figura 2)14.
Inflamosomopatías intrínsecas
Síndromes Periódicos Asociados a Criopirina (CAPS): también conocidos como Criopirinopatías, son desórdenes de una proteína esencial del Inflamosoma, denominada inicialmente como Criopirina, aunque actualmente por convención es llamada NLRP3. Las Criopirinopatías están dadas por mutaciones autosómicas dominantes o de novo, del gen NLRP3. Los CAPS agrupan a una gama de diversas patologías; éstas incluyen en orden de menor a mayor severidad al Síndrome Autoinflamatorio Familiar Inducido por Frío (FCAS), el cual se presenta con fiebre inducida por frío, rash urticariforme y síntomas constitucionales; el Síndrome de Muckle-Wells (MWS), con fiebre, pérdida auditiva neurosensorial y artritis y la Enfermedad Inflamatoria Multisistémica de Inicio Neonatal (NOMID) o Síndrome Crónico Infantil Neurológico Cutáneo y Articular (CINCA), el cual se presenta con fiebre, urticaria, sobrecrecimiento epifisiario de los huesos largos y meningitis crónica aséptica1.
El nombre de la proteína Criopirina (NLRP3), fue dado por la asociación de síntomas desencadenados por la exposición al frío en algunos pacientes (cryo) y la región invariable N-terminal de aproximadamente 90 aminoácidos, del dominio piridina. A través de este dominio, tanto la NLRP3 como la Pirina, interactúan con una proteína adaptadora denominada ASC (proteína speck-like asociada a la apoptosis). La NLRP3 pertenece a la familia de las proteínas NLR (dominio ligando de nucleótidos que contiene repeticiones ricas en leucina). Las repeticiones ricas en leucina C-terminal, han sido implicadas en la detección de componentes bacterianos, lo cual sugiere un papel de la familia NLR en la respuesta inmune innata y la activación del Inflamosoma; llevando a las investigaciones a relacionar las mutaciones del NLRP3 con alteraciones en la producción de IL-1β, en este grupo de enfermedades1,15. También ha sido señalado cómo las mutaciones del NLRP3 se saltan el requerimiento de ATP en la producción de IL-1β, evitando la necesidad de unión entre el dominio NACHT y el ATP ligando, mecanismo implicado en algunas Criopirinopatías16. Dada la sobreexpresión de IL- 1β por las mutaciones del NLRP3 en este grupo de enfermedades, se han probado con éxito medicamentos antagonistas del receptor de IL-1 como Anakinra, con importantes efectos en la mejoría clínica del NOMID/CINCA, disminuyendo el rash, la meningitis aséptica y la inflamación coclear (Figura 2)17.
Inflamosomopatías extrínsecas
Fiebre Mediterránea Familiar (FMF): está caracterizada por episodios entre 1 a 3 días, dados por fiebre con peritonitis estéril, inflamación pleural, artritis, rash y en algunas ocasiones desarrollo de amiloidosis sistémica. A pesar de que la FMF es el prototipo de enfermedad autoinflamatoria, la mutación causante de la enfermedad que afecta la producción de IL-1β puede ser primariamente extrínseca al Inflamosoma. El gen denominado MEFV que codifica para la proteína Pirina está afectado por las mutaciones que causan la FMF. Dado que en ocasiones se ha encontrado una sola mutación en el gen de la Pirina, es probable que estén implicados mecanismos de herencia poligénica. La Pirina también es conocida como TRIM20 y hace parte de una familia más grande denominada proteínas TRIpartite Motif (TRIM), implicadas en el reconocimiento de patógenos y en la regulación transcripcional de sistemas de la inmunidad innata. El fragmento Pirina N-terminal parece activar el factor nuclear kappa B (NF-κB) a través del incremento de la degradación mediada por calpaína del inhibidor kappa B-α. La Pirina también interactúa con la proteína adaptadora ASC, caracterizando posibles mecanismos moleculares implicados en la FMF, tanto inhibiendo la activación de la IL-1β compitiendo con la caspasa-1 por la ASC, como por sí misma formando un complejo inflamosomal1. Otro hallazgo interesante es cómo la Pirina interactúa con la tubulina y los microtúbulos, explicando la eficacia reportada con el uso de Colchicina en FMF (Figura 2)18.
Síndrome de Artritis Piógena con Pioderma Gangrenoso y Acné (PAPA): es un desorden autoinflamatorio hereditario de la piel y las articulaciones para el cual hay una clara evidencia de un importante pero no exclusivo papel patogénico de la IL-1β. Las familias afectadas presentan artritis recurrente, líquido sinovial inflamatorio pero estéril y manifestaciones cutáneas tales como pioderma gangrenoso y acné cístico severo. La mutación responsable de esta condición heredada de forma dominante se encuentra en la proteína-1 ligando CD2 (CD2BP1), usualmente denominada como proteína-1 interactuante con fosfatasa treonina serina prolina (PSTPIP1), que a su vez interacciona con la proteína tirosina fosfatasa PEST (PTP-PEST). Las mutaciones de PSTPIP1 disminuyen su afinidad por la PTP-PEST en el síndrome PAPA. Otros trabajos establecen cómo mutaciones de PSTPIP1 conducen a una fuerte interacción con la Pirina, produciendo disminución por el PTPPEST ligando y finalmente asociándose al síndrome PAPA por incremento en la producción de IL-1β1,19. Por otra parte, el dominio de la Pirina ligado a PSTPIP1 es un autoinhibidor de la ASC ligando y previene la formación del Piroptosoma, el cual puede promover la muerte celular y la liberación de citocinas proinflamatorias como la IL-1β en las mutaciones asociadas al síndrome PAPA20. La PSTPIP1 también se expresa en células T, sugiriendo que la enfermedad puede, en cierta forma, darse por la activación inapropiada de la respuesta inmune adaptativa (Figura 2)1.
Osteomielitis Multifocal Crónica Recurrente (CRMO), Síndrome de Majeed, Síndrome Osteítis Hiperostosis Pustulosis Acné Sinovitis (SAPHO), Deficiencia del Receptor de Antagonista de IL-1 (DIRA): el CRMO usualmente se presenta en edades tempranas de la vida (alrededor de los 10 años de edad), caracterizándose por dolor óseo, lesiones líticas en la radiografía e infiltrado inflamatorio óseo sin gérmenes con o sin fiebre. El síndrome SAPHO se expresa clínicamente por las condiciones abreviadas en su acrónimo. Tanto el CRMO como el SAPHO probablemente hacen parte de la gama de un mismo desorden. Una forma autosómica recesiva de CRMO con anemia diseritropoyética, causada por mutaciones en el gen LPIN2, es conocida como síndrome de Majeed. El LPIN2 está relacionado con la regulación de la respuesta inmune innata; por tanto, las mutaciones asociadas a LPIN2 pueden provocar un disturbio en la función protectora de los macrófagos contra el estrés oxidativo, llevando a daño tisular1,21. Otros estudios en modelos murinos han llevado a relacionar el gen PSTPIP2 con la presentación esporádica de CRMO por la prueba de desequilibrio de la transmisión1. Por otra parte, mutaciones del gen IL1RN, que codifica para el antagonista del receptor de IL-1, están asociadas a un desorden autoinflamatorio autosómico recesivo denominado como DIRA, caracterizado clínicamente por inicio neonatal de osteomielitis multifocal estéril, periostitis y pustulosis (Figura 2)22.
Síndrome de Fiebre Periódica con Hiperinmunoglobulinemia D (HIDS) y deficiencia de Cinasa Mevalonato (MKD): la enzima cinasa mevalonato es expresada ubicuamente y convierte el ácido mevalónico a ácido 5-fosfomevalónico, que cataliza un paso temprano en la vía de la biosíntesis para el colesterol e isoprenos no esteroles23. Las mutaciones en ambos alelos del gen que codifican para la enzima llevan a aciduria mevalónica, fiebre recurrente, retardo mental y muerte temprana. En el HIDS existe aproximadamente un 5% de función residual de la enzima y estos pacientes tienen fiebre recurrente, linfadenopatía, dolor abdominal y rash; posiblemente explicado porque las concentraciones tóxicas de ácido mevalónico y biosíntesis deficiente de isoprenoides llevan a perturbaciones en las vías de señalización y a apoptosis alterada de los linfocitos. Otras investigaciones han sugerido que la deficiencia de isoprenoides provocada por el trastorno en la cinasa mevalonato conduce a aumento de la actividad de la caspasa-1 e incrementa la producción de IL-1β (Figura 2)1,24.
Inflamosomopatías complejas/adquiridas
Gota/Pseudogota: son enfermedades reumáticas comunes, causadas por el depósito de cristales de urato monosódico y pirofosfato de calcio dihidratado en el tejido articular y periarticular. En tales patologías, el NLRP3 tiene un papel fundamental en el desarrollo de las complicaciones inflamatorias dadas por el depósito de microcristales, los cuales incrementan la activación de la caspasa-1 y la secreción de IL-1β de los macrófagos estimulados12. Los anteriores hallazgos son consistentes con la mejoría clínica experimentada por algunos pacientes cuando son tratados con inhibidores del receptor de IL-113.
Síndrome de Schnitzler: es una enfermedad posiblemente mediada por el sistema inmune innato, la cual está característicamente asociada a gammapatía monoclonal Ig M y en ocasiones a dolor óseo y a alteraciones radiológicas. Tiene una edad media de inicio a los 51 años y otros hallazgos que incluyen la presencia de urticaria, fiebre intermitente, artralgias, artritis, linfoadenopatía y elevación de reactantes de fase aguda, evocan a las Criopirinopatías. Las investigaciones sugieren una gran influencia de citocinas proinflamatorias del sistema inmune innato y el beneficio terapéutico de la inhibición de la IL-1β1.
Desórdenes de la activación del Factor Nuclear kappa B (NF-κB)
Síndrome Blau (BS): la condición se caracteriza por uveítis granulomatosa, artritis, rash y camptodactilia. El síndrome Blau está dado por mutaciones en el NOD2 heredadas dominantemente, cuyo blanco es el dominio NBD/NACHT de la proteína NLRP3, que resultan en la sobreactivación del NF-κB y posiblemente en alteración del procesamiento de ATP (Figura 2)1,25.
Desórdenes en el plegamiento de proteínas en el Sistema Inmune Innato
Síndrome Periódico Asociado al Receptor TNF (TRAPS): es producido por una mutación en el gen TNFRSF1A que codifica para el receptor TNF-1 y es heredada dominantemente. Los pacientes con TRAPS experimentan episodios febriles prolongados, dolor abdominal severo, pleuritis, artritis, rash migratorio con fascitis subyacente, edema periorbitario y eventualmente desarrollan amiloidosis sistémica. Las mutaciones del TNFRSF1A asociadas al TRAPS afectan la región extracelular del receptor y en algunos casos los residuos de cisteína que participan en los enlaces disulfuro, resultando en un fenotipo con alto riesgo de amiloidosis26. Las mutaciones pueden llevar a un defecto en la habilidad de las metaloproteinasas para clivar el TNFR1 de la membrana celular, disminuyendo la inactivación del receptor y permitiendo el estímulo repetido sobre el mismo. Otros datos sugieren que la acumulación de TNFR1 mutante resulta en la activación espontánea de las MAPK, llevando a las células a ser más susceptibles a los estímulos inflamatorios; tanto así que algunos pacientes con TRAPS se han beneficiado con la administración de un anti-TNF como la proteína dimérica de fusión Etanercept1.
Referencias
1. Masters S, Simon A, Aksentijevich I, Kastner D. Horror Autoinflammaticus: The Molecular Pathophysiology of Autoinflammatory Disease. Annu Rev Immunol 2009;27:621-668. [ Links ]
2. Harton JA, Linhoff MW, Zhang J, Ting JP. Cutting edge: CATERPILLER: a large family of mammalian genes containing CARD, pyrin, nucleotide-binding, and leucine-rich repeat domains. J Immunol 2002;169(8):4088-4093. [ Links ]
3. Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet 2001;29:301-305. [ Links ]
4. Tschopp J, Martinon F, Burns K. NALPs: a novel protein family involved in inflammation. Nature Reviews Molecular Cell Biology 2003;(4):95-104. [ Links ]
5. Franchi L, Eigenbrod T, Muñoz-Planillo R, Núñez G. The inflammasome: a caspase-1 activation platform that regulates immune responses and disease pathogenesis. Nat Immunol 2009;10:241-247. [ Links ]
6. Eicke L. The inflammasomes: mechanisms of activation and function. Current Opinion in Immunology 2010;22:28-33. [ Links ]
7. Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell 2004;117:561-574. [ Links ]
8. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 2002;10:417-426. [ Links ]
9. Miao EA, et al. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1β via Ipaf. Nat Immunol 2006;7:569-575.
10. Mariathasan S, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006;440:228-232. [ Links ]
11. Pétrilli V, Dostert C, Muruve DA, Tschopp J. The inflammasome: a danger sensing complex triggering innate immunity. Current Opinion in Immunology 2007;19:615-622. [ Links ]
12. Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006;440:237-241. [ Links ]
13. So A, De Smedt T, Revaz S, Tschopp J. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res Ther 2007;9(2):R28. [ Links ]
14. Pétrilli V, Martinon F. The inflammasome, autoinflammatory diseases, and gout. Joint Bone Spine 2007;74:571-576. [ Links ]
15. Aksentijevich I, Nowak M, Mallah M, Chae JJ, Watford WT, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum 2002;46:3340-3348. [ Links ]
16. Duncan JA, Bergstralh DT, Wang Y, Willingham SB, Ye Z, Zimmermann AG, Ting JP. Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc Natl Acad Sci USA 2007;104:8041-8046. [ Links ]
17. Goldbach-Mansky R, Dailey NJ, Canna SW, Gelabert A, Jones J, et al. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1β inhibition. N Engl J Med 2006;355:581-592.
18. Mansfield E, Chae JJ, Komarow HD, Brotz TM, Frucht DM, et al. The familial Mediterranean fever protein, pyrin, associates with microtubules and colocalizes with actin filaments. Blood 2001;98:851-859. [ Links ]
19. Shoham NG, Centola M, Mansfield E, Hull KM, Wood G, et al. Pyrin binds the PSTPIP1/ CD2BP1 protein, defining familial Mediterranean fever and PAPA syndrome as disorders in the same pathway. Proc Natl Acad Sci USA 2003;100:13501-13506. [ Links ]
20. Yu JW, Fernandes-Alnemri T, Datta P, Wu J, Juliana C, et al. Pyrin activates the ASC pyroptosome in response to engagement by autoinflammatory PSTPIP1 mutants. Mol Cell 2007;28:214-227. [ Links ]
21. El-Shanti HI, Ferguson PJ. Chronic recurrent multifocal osteomyelitis: a concise review and genetic update. Clin Orthop Relat Res 2007;462:19. [ Links ]
22. Aksentijevich I, Masters S, Ferguson P, et al. An Autoinflammatory Disease with Deficiency of the Interleukin-1-Receptor Antagonist. N Engl J Med 2009;360:2426-2437. [ Links ]
23. Hoffmann G, Gibson KM, Brandt IK, Bader PI, Wappner RS, Sweetman L. Mevalonic aciduria-an inborn error of cholesterol and nonsterol isoprene biosynthesis. N Engl J Med 1986;314:1610-1614. [ Links ]
24. Kuijk LM, Beekman JM, Koster J, Waterham HR, Frenkel J, Coffer PJ. HMG-CoA reductase inhibition induces IL-1β release through Rac1/PI3K/PKBdependent caspase-1 activation. Blood 2008; 112:3563-3573.
25. Kanazawa N, Okafuji I, Kambe N, Nishikomori R, Nakata-Hizume M, et al. Early-onset sarcoidosis and CARD15 mutations with constitutive nuclear factor-κB activation: common genetic etiology with Blau syndrome. Blood 2005;105:1195-1197.
26. Aksentijevich I, Galon J, Soares M, Mansfield E, Hull K, et al. The tumor-necrosis-factor receptor associated periodic syndrome: new mutations in TNFRSF1A, ancestral origins, genotype-phenotype studies, and evidence for further genetic heterogeneity of periodic fevers. Am J Hum Genet 2001;69:301-314. [ Links ]

