










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Colombiana de Reumatología
versión impresa ISSN 0121-8123
Rev.Colomb.Reumatol. v.17 n.4 Bogotá sep./dic. 2010
PRESENTACIÓN DE CASO Y REVISIÓN DE LA LITERATURA
1 Residente tercer año Medicina Interna. Facultad de Medicina. Universidad Nacional de Colombia.
2 Médico Interno. Universidad Nacional de Colombia.
3 Reumatólogo. Profesor Asociado de Medicina Interna y Reumatología. Universidad Nacional de Colombia.
4 Reumatólogo. Profesor Titular de Medicina Interna y Reumatología. Universidad Nacional de Colombia.
Correspondencia: Antonio Iglesias Gamarra iglesias.antonio1@gmail.com
Recibido: 22 de octubre de 2010. Aceptado: 6 de diciembre de 2010.
Resumen
La osteomalacia inducida por tumor es un síndrome paraneoplásico secundario en la mayoría de los casos a tumores de origen mesenquimal. Se caracteriza por pérdida aumentada de fosfato a nivel urinario por el efecto inhibidor que ejerce el factor de crecimiento fibroblástico 23 sobre el transporte de fósforo en el túbulo renal proximal. Debe sospecharse en un paciente con debilidad y dolor osteomuscular generalizado que se presente con hipofosfatemia, normocalcemia, fosfatasa alcalina elevada y niveles de 25 hidroxivitamina D y PTH normales. El tratamiento definitivo de la enfermedad es la resección quirúrgica del tumor. Cuando se desconozca la neoplasia primaria o no sea posible el tratamiento quirúrgico debe iniciarse reposición de fósforo y calcitriol. En este artículo se presenta el primer caso de una paciente con osteomalacia inducida por tumor asociada a un carcinoma lobulillar infiltrante de seno.
Palabras clave: osteomalacia, hipofosfatemia, 25-hidroxivitamina D, parathormona (PTH).
Summary
The tumor-induced osteomalacia is a paraneoplastic syndrome secondary in most cases to tumors of mesenchymal origin. It is characterized by increased lost of urinary phosphate by the inhibitory effect exerted by the fibroblast growth factor 23 on phosphorus transport in the proximal renal tubule. Should be suspected in a patient with weakness and generalized muscle in addition to hypophosphatemia, normocalcemia, elevated alkaline phosphatase and normal serum 25-hydroxyvitamin D and PTH. The definitive treatment of the disease is surgical resection of the tumor. When the primary tumor is unknown or is not possible the surgical treatment should be initiated replacement of phosphorus and calcitriol. This paper presents the first case of a patient with tumor-induced osteomalacia associated with lobular breast cancer.
Key words: osteomalacia, hypophosphatemia, 25-hydroxyvitamin D, parathormone(PTH).
Introducción
La osteomalacia inducida por tumor (OIT), también conocida como osteomalacia oncogénica, es una entidad rara clasificada dentro del grupo de síndromes paraneoplásicos. Se caracteriza por una disminución en la reabsorción de fosfato a nivel tubular, que da lugar a hipofosfatemia marcada, hiperfosfaturia y alteraciones en la mineralización; dichas alteraciones en el metabolismo óseo se constituyen entonces en manifestaciones subyacentes de la presencia de un tumor usualmente de origen mesenquimal. En este informe se presenta el caso de una paciente con osteomalacia inducida por un carcinoma lobulillar infiltrante de seno.
Caso clínico
Paciente de 43 años sin antecedentes médicos de importancia que consulta por cuadro de un año de evolución consistente en pérdida de peso no cuantificada y aparición progresiva de masas de en ambos senos. Se ordena mamografía, la cual informa lesiones sospechosas de malignidad por lo cual se decide llevar a estudio histopatológico. El diagnóstico definitivo revela un carcinoma lobulillar infiltrante grado nuclear 1 con compromiso cutáneo. Posterior a la estadificación, es valorada por servicio de oncología, quienes inician quimioterapia con ciclofosfamida, doxorrubicina, 5-fluoracilo, dexametasona y programan para mastectomía radical modificada bilateral más vaciamiento ganglionar. De manera concomitante con la aparición del tumor, la paciente refiere dolor osteomuscular generalizado, artralgias, debilidad y fatiga; dichos síntomas motivan la remisión al servicio de reumatología. Al momento de la valoración, se palpan masas de consistencia pétrea en mama derecha, sin signos objetivos de compromiso cutáneo y adenopatías axilares de aproximadamente un centímetro. Al examen osteoarticular no se encuentra evidencia de sinovitis, ni dolor a la palpación articular. El único hallazgo positivo es ligera disminución de la fuerza muscular a nivel proximal en miembros inferiores.
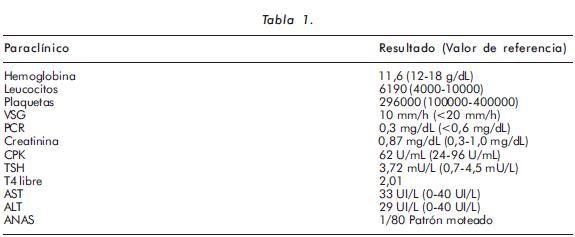
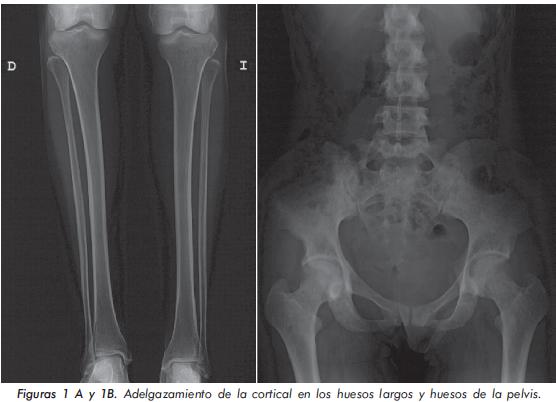
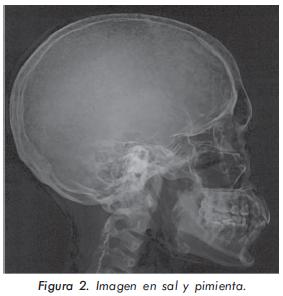
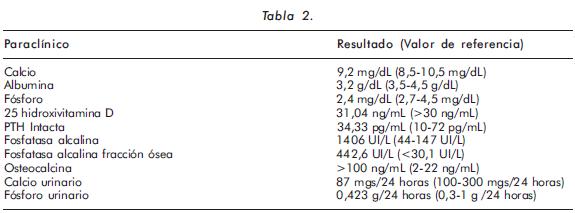
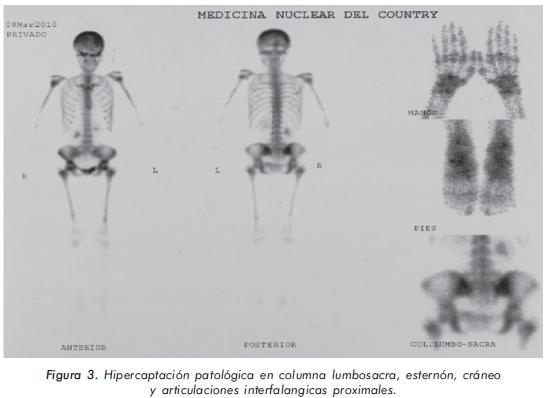
Se le solicitan estudios en búsqueda de causas metabólicas o asociadas al tumor (metástasis óseas) que expliquen el dolor osteomuscular generalizado. La química sanguínea solicitada inicialmente muestra una anemia leve, con función renal, hepática y tiroidea dentro de límites normales. Reactantes de fase aguda, CPK, ELISA VIH y ANAS negativos (Tabla 1). En la radiografía de columna lumbosacra llama la atención osteopenia en cuerpos vertebrales, sin lesiones metastásicas ni fracturas por aplastamiento. En las de huesos largos y pelvis se observa un marcado adelgazamiento de la cortical (Figuras 1A y 1B). La radiografía de cráneo documentó imagen en sal y pimienta (Figura 2). En ese contexto, se decide ampliar estudios para descartar compromiso tumoral a hueso y evaluar metabolismo mineral óseo, documentándose hipofosfatemia, normocalcemia, fosfatasa alcalina total-ósea y osteocalcina elevadas con niveles de 25(OH)D3 y PTH normales (Tabla 2). La gammagrafía ósea muestra hipercaptación patológica en columna lumbosacra, esternón, cráneo y articulaciones interfalángicas proximales (Figura 3). La densitometría ósea revela osteopenia en columna lumbar y cuello femoral.
Se considera que el cuadro clínico, los resultados del perfil bioquímico y las anormalidades observadas tanto en la radiología simple como en la gamagrafía ósea son compatibles con osteomalacia hipofosfatémica. A pesar de que el fósforo urinario no se encuentra en un valor absoluto disminuido, si está cerca del límite inferior bajo. Al realizarse este diagnóstico debe establecerse su causa entre varias patologías: osteomalacia hipofosfatémica ligada al cromosoma X, osteomalacia hipofosfatémica esporádica y osteomalacia inducida por tumor.
La primera posibilidad, en vista de la carencia de antecedentes familiares, queda descartada. También quedan descartados baja ingesta de fosfatos, ingesta de antiácidos ligadores de fosfatos, displasia fibrosa del hueso, neurofibromatosis, síndrome nevus sebáceo linear y nefropatía de cadenas livianas, que se han descrito como causas adquiridas de osteomalacia hipofosfatémica. De esta manera, se hace diagnóstico de osteomalacia inducida por tumor, asociada a carcinoma lobulillar infiltrante de seno.
La paciente fue sometida a mastectomía radical modificada bilateral más quimioterapia neoadyuvante y adyuvante. Posterior a la resección del tumor los síntomas músculo-esqueléticos desaparecieron, lo cual apoya el diagnóstico de osteomalacia inducida por tumor.
Discusión
La OIT es un síndrome infrecuente que se caracteriza por alteraciones en el metabolismo mineral producidas por la presencia de tumores fosfatúricos. Aunque McCrance1 describió el primer caso de OIT en 1947, Prader, et al.2 fueron los primeros en reconocer la asociación causal del síndrome en 1959. A la fecha se han descrito aproximadamente 130 casos en los cuales se ha podido determinar la relación entre osteomalacia y tumor, encontrándose en un buen porcentaje de los casos la resolución del trastorno metabólico con la remoción quirúrgica del tumor.
A pesar de su baja incidencia, el progreso en el conocimiento de su patogénesis ha contribuido de manera importante al entendimiento de la homeostasis del fósforo corporal y a comprender mejor la amplia gama de trastornos hipofosfatémicos.
Manifestaciones clínicas
Aunque la mayoría de pacientes con OIT son adultos mayores, el síndrome puede manifestarse a cualquier edad. La edad promedio al momento del diagnóstico es la sexta década de la vida con un rango entre 7 y 74 años3. La duración promedio de los síntomas antes del diagnóstico oscila entre 2,5 meses y 19 años con un promedio de 2,5 años4. Usualmente se presenta con síntomas inespecíficos de larga evolución que incluye dolor osteomuscular generalizado, debilidad muscular, fatiga y menos frecuente fracturas patológicas de huesos largos. En la mayoría de los casos el tumor subyacente no ha producido manifestaciones clínicas al momento de inicio de los síntomas de OIT. Los niños con OIT usualmente presentan rasgos clásicos de raquitismo como alteraciones en la marcha, retardo en el crecimiento y deformidades esqueléticas.
Las alteraciones bioquímicas del trastorno incluyen bajas concentraciones de fósforo, fosfaturia secundaria a una reducida reabsorción tubular proximal de fosfato y unos bajos o inapropiados niveles normales de 1,25(OH)2D que deberían encontrarse altos como respuesta fisiológica compensadora a la hipofosfatemia. La PTH se encuentra elevada tan solo en un pequeño porcentaje de pacientes y la fosfatasa alcalina típicamente se encuentra aumentada. No es infrecuente observar otras alteraciones en la reabsorción tubular como glicosuria y aminociduria5.
Evaluación diagnóstica
El diagnóstico de la enfermedad es a menudo un reto dada la inespecificidad de las manifestaciones clínicas y requiere una cuidadosa evaluación con estudios de laboratorio e imágenes radiológicas. El tamizaje metabólico debe incluir fósforo sérico en ayunas, calcemia, fosfatasa alcalina, PTH, creatinina e idealmente niveles de 1,25(OH)2D. Además debe evaluarse concentraciones en orina de fósforo, calcio, creatinina, glucosa y aminoácidos. Como la principal característica de la enfermedad es la alteración en la reabsorción de fosfato, el cociente reabsorción tubular máxima de fosfato/tasa de filtración glomerular (TmP/GFR) debe ser calculado6.
Las alteraciones radiográficas incluyen osteopenia generalizada, seudofracturas, así como discos epifisiarios ensanchados en niños. En algunos casos, para confirmar el diagnóstico histológico una biopsia de la cresta iliaca con tetraciclina doblemente marcada es obtenida para realizar estudios histomorfométricos de hueso7.
La detección y localización del tumor desencadenante de OIT es obligatoria ya que la resección quirúrgica completa es curativa. Sin embargo, los tumores mesenquimales que causan OIT son generalmente pequeños, de crecimiento lento y se encuentran en sitios anatómicos poco frecuentes, por lo que las imágenes tradicionales usualmente no detectan el tumor. La localización de este tipo de tumores debe realizarse, entonces, con técnicas que utilicen marcadores con afinidad por los receptores tumorales. Se ha demostrado in vitro que muchos tumores mesenquimales expresan receptores de somatostatina8. La gamagrafía con octreotide, una técnica que usa un análogo de somatostatina como radiomarcador, ha demostrado ser muy útil en la localización del tumor mesenquimal9; así mismo se ha descrito que la resonancia magnética corporal total10 y la tomografía con emisión de positrones con flúor 18 (F18) unido a fluordeoxiglucosa (FDG)11 son imágenes sensibles en el diagnóstico del tumor y deben ser utilizadas principalmente en región craneofacial y extremidades dada la mayor prevalencia de tumores mesenquimales en dicha localización12.
Estudios recientes han encontrado que el factor de crecimiento fibroblástico 23 (FGF-23) es una sustancia fosfatúrica producida por tumores que inducen osteomalacia13. Los genes que promueven la expresión de FGF-23 se encuentran aumentados en OIT. El efecto de inhibir el transporte de fósforo a través del epitelio tubular renal proximal ha sido demostrado in vitro14, y la reducción del fósforo sérico y el incremento en la fracción excretada de fósforo ha sido demostrada en ratones15. Niveles de FGF-23 se han detectado tanto en pacientes con OIT como en pacientes con displasia fibrosa16 e hipofosfatemia ligada al cromosoma X17; así mismo se ha encontrado en insuficiencia renal y se correlaciona positivamente con la concentración de fosfato y creatinina sérica18,19. Su valor diagnóstico en OIT se ha confirmado al encontrarse disminución en los niveles séricos con la resección del tumor20.
Se han comparado distintos métodos para evaluar la concentración sérica de FGF-23 en OIT. La mayor sensibilidad se encontró con la técnica de inmunoensayo de kainos, la cual detectó niveles de FGF-23 en el 86% de pacientes con OIT. Los pacientes cuyo tumor ha sido localizado tienen mayor probabilidad de presentar elevación sérica de FGF-23, esto podría deberse a un incremento en la producción de FGF-23 proporcional al tamaño del tumor. En los casos en los cuales se ha localizado el tumor la técnica de kainos tiene una sensibilidad del 100%21.
El hecho de que algunos pacientes no tengan elevado el FGF-23 podría indicar que existen limitaciones en los métodos disponibles para evaluar sus niveles séricos. Además, en algunos casos, el cuadro clínico puede no ser causado por aumento en FGF-23 y algunos tumores podrían expresar otras sustancias responsables de la osteomalacia y la excreción de fosfatos. De Beur, et al.22, demostraron que los OIT tienen una sobreexpresión de una fosfoglicoproteína de la matriz extracelular denominada sFRP4. Esta proteína hace parte del grupo de las fosfatoninas y actúa al inhibir la vía Wnt. Esta cascada de señalización es importante en la homeostasis de la mineralización ósea. Algunas evidencias experimentales sugieren que sFRP4 tiene propiedades fosfatúricas. sFRP4 inhibe el transporte de fosfato en células epiteliales renales e induce hipofosfatemia y alteración en los mecanismos de autorregulación del 1,25(OH)2D23. Otras fosfatoninas que se han implicado en la patogenésis de la enfermedad son el factor de crecimiento fibroblástico 7 (FGF-7)24 y la fosfoglicoproteína de matriz extracelular (MEPE)25. Ambas han demostrado en cultivos celulares tener actividad fosfatúrica.
Tumores causantes de OIT
Los tumores descritos en pacientes con OIT han sido de origen mesenquimal en la mayoría de los casos. Sin embargo, la presencia de nodos de Looser en radiografías de pacientes terminales con carcinomas derivados del tejido endo y ectodérmico indica que la enfermedad puede ser secundaria a un amplio espectro de tumores26. Esta conclusión la confirman los casos publicados en la literatura de OIT secundarios a carcinoma (Ca) de seno, Ca de Próstata, Ca Renal de células claras, mieloma múltiple y leucemia linfoide crónica4. Específicamente en relación con el cáncer de seno se ha publicado en la literatura la ocurrencia de osteomalacia en pacientes con displasia fibrosa ósea, neurofibromatosis, y el síndrome de nevus sebáceo linear podría así mismo ser considerado como inducido por tumor, a pesar de que una relación de causalidad sea difícil de comprobar al tratarse de patologías de difícil tratamiento quirúrgico dado el compromiso extenso de la superficie corporal.
Los tumores mesenquimales asociados con OIT son angioma esclerosante, angiofibroma benigno, hemangiopericitoma, condrosarcoma, tumor mesenquimal primitivo, tumor de partes blandas similar a condroma y el tumor de células gigantes óseo. En la mayoría de los casos son pequeños, indolentes y difíciles de localizar. Aunque se han encontrado en diferentes regiones anatómicas incluyendo nasofaringe, senos paranasales y la ingle, su localización más frecuente son las extremidades y el esqueleto apendicular27.
La variedad de diagnósticos morfológicos pone de manifiesto la complejidad de este tipo de tumores. Sin embargo, Weidner y Cruz28 han establecido con base en el patrón histológico una clasificación con cuatro tipos de patrones: 1) tumor mesenquimal fosfatúrico tipo tumor de tejido conectivo mixto (PMTMCT), 2) tumor similar a osteoblastoma, 3) tumor no osificante similar a fibroma y 4) tumor osificante similar a fibroma. La variedad más común es PMTMCT, se presenta en tejidos blandos, su comportamiento usualmente es benigno y se caracteriza por un número variable de células estromales primitivas de crecimiento pobremente definido en agrupaciones de células gigantes similares a osteoclastos29. Probablemente las células primitivas estromales primitivas sean la fuente de los factores hormonales causantes del síndrome; sin embargo, esta hipótesis no se ha podido comprobar.
Tratamiento
El tratamiento definitivo de la enfermedad es la resección quirúrgica del tumor. Esto produce rápidamente corrección de las alteraciones bioquímicas características de los OIT y remineralización ósea. Sin embargo, incluso después que el diagnóstico de OIT es realizado, en algunas ocasiones el tumor no se logra identificar o es imposible su resección total por lo cual el manejo médico de la entidad es necesario en muchos casos.
El tratamiento actual de OIT incluye la suplencia de fósforo para remplazar las pérdidas renales y la administración de calcitriol para compensar la insuficiente producción renal de 1,25(OH)2D y elevar la reabsorción gastrointestinal y tubular de fósforo. Usualmente las dosis utilizadas son 2 gramos/día de fósforo y 1-3 ucg de calcitriol6. En algunos casos la administración única de calcitriol puede mejorar las anormalidades bioquímicas de la OIT y curar la osteomalacia. Los objetivos terapéuticos son mejorar los síntomas, mantener los niveles séricos de fósforo en ayunas en un rango normal bajo, normalizar la fosfatasa alcalina y controlar el hiperparatiroidismo secundario sin inducir hipercalcemia o hipercalciuria.
Es necesario monitorizar las posibles complicaciones de las altas dosis de calcitriol y fósforo como hipercalcemia, nefrocalcinosis y nefrolitiasis. Así mismo se debe monitorizar la función paratiroidea y la suplencia de fósforo (solo o en combinación con vitamina D) ya que esta puede estimular la función paratiroidea y eventualmente producir autonomía (hiperparatiroidismo terciario). Se recomienda monitorizar la eficacia y seguridad del tratamiento con calcio urinario y sérico, fósforo sérico, función renal y PTH cada tres meses6.
Seufert, et al.30 sugirieron que la terapia con octreotide puede ser un tratamiento alternativo para OIT. Se han encontrado receptores de somatostatina en tumores mesenquimales y su uso se fundamenta en la inhibición de la secreción hormonal y la regulación del metabolismo del fósforo. La terapia con octreotide normaliza el perfil bioquímico y mejora la sintomatología producida por OIT.
Referencias
1. McCance RA. Osteomalacia with Looser's nodes due to a raised resistance to vitamin D acquired about the age of 15 years. Q J Med 1947;16:33-46. [ Links ]
2. Prader V, Illig R, Vehlinger E, Stalder G. Rachitis, infolge, knochen tumors. Helv Pa ediatr Acta 1959;14:554-565. [ Links ]
3. Drezner MK. Tumor-induced osteomalacia. In: Favus MJ (ed.) Primer on the metabolic bone diseases and disorders of mineral metabolism, 4th ed. LippincottRaven, Philadelphia, PA, USA, 1999;331-337. [ Links ]
4. Drezner MK.Tumor-induced osteomalacia. Rev Endocr Metab Disord 2001;2:175-186. [ Links ]
5. Kumar R. Tumor-induced osteomalacia and the regulation of phosphate homeostasis. Bone 2000; 27:333-338. [ Links ]
6. Jan De Beur SM. Tumor-induced osteomalacia. In: Favus MJ (ed.) Primer on the metabolic bone diseases and disorders of mineral metabolism, 6th ed. LippincottRaven, Philadelphia, PA, USA, 2006;345-350. [ Links ]
7. Weiss D, Bar RS, Weidner N. Oncogenic osteomalacia. Ann Intern Med 198;102:557. [ Links ]
8. Reubi J C, Waser B, Laissue JA, Gebbers JO. Somatostatin and vasoactive intestinal peptide receptors in human mesenchymal tumors: in vitro identification. Cancer Res 1996;56:1922-1931. [ Links ]
9. Garcia C, Spencer R. Bone and In-111 octreotide imaging in oncogenic osteomalacia: a case report. Clin Nucl Med 2002;27:582-583. [ Links ]
10. Avila NA, Skarulis M, Rubino DM, Doppman JL. Oncogenic osteomalacia: lesion detection by MR skeletal survey. Am J Roentgenol 1996;167:343-345. [ Links ]
11. Dupond JL, Mahammedi H, Prié D. Oncogenic osteomalacia: diagnostic importance of fibroblast growth factor 23 and F-18 fluorodeoxyglucose PET/CT scan for the diagnosis and follow-up in one case. Bone 2005;36:375-378. [ Links ]
12. Jan de Beur SM. Tumor-induced osteomalacia. JAMA 2005;294:1260-1267. [ Links ]
13. Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, et al. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci USA 2001;98:6500-6505. [ Links ]
14. Yamashita T, Konsishi M, Miayke A. Fibroblast growth factor (FGF)-23 inhibits renal phosphate reabsorption by activation of themitogen-activated protein kinase pathway. J Biol Chem 2002;277:28265-28270. [ Links ]
15. Shimada T, Muto T, Urakawa I, Yoneya T, Yamazaki Y, Okawa K, et al. Mutant FGF-23 responsible for autosomal dominant hypophosphatemic rickets is resistant to proteolytic cleavage and causes hypophosphatemia in vivo. Endocrinology 143:3179-3182. [ Links ]
16. Riminucci M, Collins MT, Fedarko NS, Cherman N, Corsi A, White KE, et al. FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J Clin Invest 2003;112:683-692. [ Links ]
17. Nelson AE, Bligh RC, Mirams M, Gill A, Au A, Clarkson A, et al. Fibroblast growth factor 23: a new clinical marker for oncogenic osteomalacia. J Clin Endocrinol Metab 2003;88:4088-4094. [ Links ]
18. Larsson T, Nisbeth U, Ljunggren O, Juppner H, Jonsson KB. Circulating concentration of FGF-23 increases as renal function declines in patients with chronic kidney disease, but does not change in response to variation in phosphate intake in healthy volunteers. Kidney Int 2003;64:2272-2279. [ Links ]
19. Imanishi Y, Inaba M, Nakatsuka K, Nagasue K, Okuno S, Yoshihara A, et al. FGF-23 in patients with end-stage renal disease on hemodialysis. Kidney Int 2004;65:943-946. [ Links ]
20. Takeuchi Y, Suzuki H, Ogura S, Imai R, Yamazaki Y, Yamashita T, et al. Venous sampling for fibroblast growth factor-23 confirms preoperative diagnosis of tumorinduced osteomalacia. J Clin Endocrinol Metab 2004;89:3979-3982. [ Links ]
21. Imel EA, Peacock M, Pitukcheewanont P, Heller HJ, Ward LM, Shulman D, et al. Sensitivity of fibroblast growth factor 23 measurements in tumor-induced osteomalacia. J Clin Endocrinol Metab 2006;91: 2055-2061. [ Links ]
22. Jan De Beur SM, Finnegan RB, Vassiliadis J, Cook B, Barberio D, Estes S, et al. Tumors associated with oncogenic osteomalacia express genes important in bone and mineral metabolism. J Bone Miner Res 2002;17:1102-1110. [ Links ]
23. Berndt T, Craig TA, Bowe AE, Vassiliadis J, Reczek D, Finnegan R, et al. Secreted frizzled-related protein 4 is a potent tumor-derived phosphaturic agent. J Clin Invest 2003;112:785-794. [ Links ]
24. Carpenter TO, Ellis BK, Insogna KL, Philbrick WM, Sterpka J, Shimkets R. FGF7, an inhibitor of phosphate transport derived from oncogenic osteomalaciacausing tumors. J Clin Endocrinol Metab 2005;90:1012-1020. [ Links ]
25. Rowe PS, de Zoysa PA, Dong R, Wang HR, White KE, Econs MJ, et al. MEPE, a new gene expressed in bone marrow and tumors causing osteomalacia. Genomics. 2000;67:54-68. [ Links ]
26. Dent CE, Stamp TCB. Vitamin D rickets and osteomalacia. In: Avioli LV, Krane S, eds. Metabolic Bone Disease, vol 1. New York: Academic Press, 1978;237. [ Links ]
27. Whyte MP, Podgornik MN, Wollberg VA, Eddy MC, McAlister WH. Pseudo-(tumor-induced) rickets. J Bone Miner Res. 2001;16:1564-1571. [ Links ]
28. Weidner N, Cruz DS. Phosphaturic mesenchymal tumors: a polymorphous group causing osteomalacia or rickets. Cancer 1987;59:1442-1454. [ Links ]
29. Folpe AL, Fanburg-Smith JC, Billings SD, Bisceglia M, Bertoni F, Cho JY, et al. Most osteomalaciaassociated mesenchymal tumors are a single histopathologic entity: an analysis of 32 cases and a comprehensive review of the literature. Am J Surg Pathol 2004;28:1-30. [ Links ]
30. Seufert J, Ebert K, Müller J, Eulert J, Hendrich C, Werner, et al. Octreotide therapy for tumor-induced osteomalacia. N Engl J Med 2001;345:1883-1888. [ Links ]

