










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Reumatología
Print version ISSN 0121-8123
Rev.Colomb.Reumatol. vol.17 no.4 Bogotá Sept./Dec. 2010
PRESENTACIÓN DE CASO Y REVISIÓN DE LA LITERATURA
1Residente de Medicina Interna, Universidad Pontificia Bolivariana.
2Internista Reumatólogo, Hospital Pablo Tobón Uribe, Universidad Pontificia Bolivariana, Medellín, Colombia.
3Neumólogo Clínica Santa María, Centro Cardiovascular Colombiano, Medellín, Colombia.
Correspondencia: Luis Fernando Pinto: lfpintop@hotmail.com
Recibido: 18 de noviembre de 2010. Aceptado: 20 de diciembre de 2010.
Resumen
Se reporta el caso de un paciente que presentó un síndrome antisintetasa en el cual la miopatía fue precedida por EPID grave de instalación rápida y buena respuesta al manejo inmunosupresor con corticosteroides, ciclofosfamida y azatioprina. El cuadro clínico inicial con fiebre, infiltrados pulmonares, SDRA y ausencia de miopatía fue muy sugestivo de infección.
Palabras clave: miopatía inflamatoria, enfermedad pulmonar intersticial, síndrome antisintetasa.
Summary
We report a patient with antisintetase síndrome with rapid and progressive interstitial difuse pulmonary disease preceding the muscular weakness. The patient was successfully treated with steroids, cyclophosphamide and azathioprine. The initial compromise: fever, dispnoea and pulmonary infiltrates without miopathy was misdiagnosed as pneumonia.
Key words: inflamatory miopathy, interstitial pulmonary disease, antisintetase syndrome.
Las miopatías inflamatorias conforman el principal grupo de miopatías adquiridas del adulto1. En este se incluyen: la dermatomiositis, la polimiositis y la miositis de cuerpos de inclusión2, los cuales tienen una incidencia aproximada de 0,6 a 1 por 100.000 personas1. Estas enfermedades se caracterizan por debilidad muscular simétrica y proximal de instauración insidiosa, usualmente no dolorosa1, con elevación de las enzimas musculares hasta 50 veces por encima de su valor normal, alteraciones evidentes en la biopsia de músculo en la microscopía electrónica y de luz y por cambios electromiográficos característicos3. El carácter autoinmune de estas enfermedades se ve demostrado por la presencia de infiltrado inflamatorio en el tejido muscular y por la presencia de diversos autoanticuerpos ente los cuales se destacan: Anti-SRP (que predicen compromiso cardíaco en la enfermedad)4, Anti-Mi2 (que identifica un subgrupo de dermatomiositis con respuesta adecuada a esteroides) y los anticuerpos antisintetasa (que predicen compromiso pulmonar asociado a la enfermedad intersticial).
Presentación del caso
Paciente de 48 años previamente sano quien ocho meses antes presentó un cuadro de fiebre, astenia y tos seca seguido por disnea que progresó rápidamente hasta la falla respiratoria, por lo que requirió ventilación mecánica; por la presencia de infiltrados difusos en ambos campos pulmonares se manejó como SDRA secundario a neumonía adquirida en la comunidad; se cubrió con antibióticos para gérmenes comunes y, ante la pobre respuesta del paciente y de una prueba presuntiva positiva para virus de inmunodeficiencia humana (VIH), se inició manejo empírico para Pneumocystis Jiroveci con antibióticos y corticosteroides. El paciente evolucionó satisfactoriamente y se descartó la infección por VIH con una prueba confirmatoria (Western Blot).
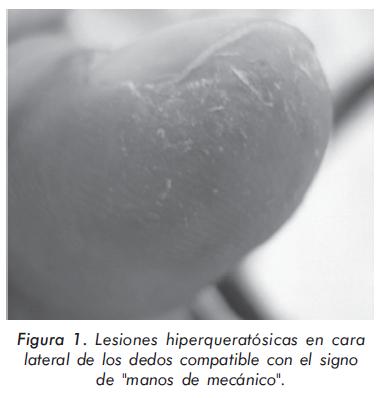
Ambulatoriamente grupo de Neumología inició tratamiento con prednisolona 1 mg/Kg/día y azatioprina 2 mg/Kg/día con diagnóstico de enfermedad pulmonar intersticial (EPID) tipo neumonía intersticial usual; la evolución clínica inicial del paciente fue satisfactoria pero al iniciar el desmonte de los esteroides presentó debilidad muscular progresiva que no mejoró con la disminución de estos medicamentos, con impresión diagnóstica de miopatía esteroidea. Al examen físico se encontró debilidad muscular de los flexores del cuello (4/5) y de las cinturas escapular (3/5) y pélvica (1/5); además se demostraron crépitos secos en ambas bases pulmonares y lesiones digitales descamativas sugestivas de "manos de mecánico" (Figura 1).
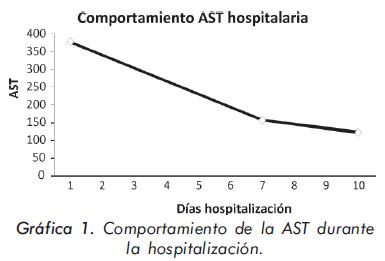
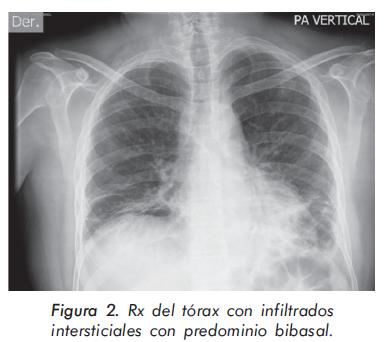
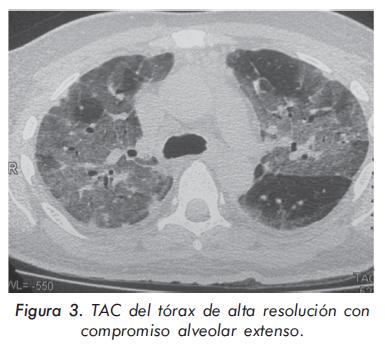
La evolución de las enzimas musculares se describe en las Gráficas 1 y 2 y la evolución radiológica en las Figuras 2 a 4; la electromiografía mostró una miopatía inflamatoria, los anticuerpos antinucleares fueron positivos con títulos de 1:640 y patrón moteado fino, los anticuerpos antiJo-1 fueron positivos. La tomografía del tórax de alta resolución evidenció un patrón de vidrio despulido sugestivo de alveolitis y panalización bibasal compatible con fibrosis pulmonar. La biopsia de músculo mostró cambios inespecíficos con degeneración y regeneración de las fibras musculares y expresión de complejo mayor de histocompatibilidad tipo I; se consideró que los hallazgos histológicos estaban modificados porque la muestra fue tomada cuando el paciente llevaba varios meses de manejo con esteroides.
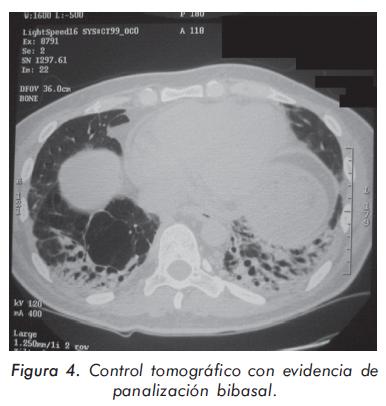
El paciente fue tratado con tres pulsos de metilprednisolona de 1 gramo/día, prednisolona 1 mg/Kg/día, en forma consecutiva, y ciclofosfamida pulsos de 1 gramo/mes por seis meses seguidos de azatioprina 2 mg/Kg/día. La respuesta al manejo fue lenta y progresiva; en la actualidad se encuentra en clase funcional II, no presenta tos ni disnea y la fuerza muscular es de 4/5 en la cintura pélvica y de 5/5 en la cintura escapular y en los músculos flexores del cuello; recibe 2 mg/Kg/día de azatioprina y 15 mg/día de prednisolona. El diagnóstico definitivo es síndrome antisintetasa que debutó con EPID, antes de presentar manifestaciones más específicas como miopatía y "manos de mecánico" que sólo fueron evidentes después del descenso en las dosis de corticosteroides.
Discusión
Las miopatías inflamatorias idiopáticas incluyen un grupo de entidades amplio y complejo de manera que clasificarlas como dermatomiositis y polimiositis parece ser insuficiente y simplista; hay evidencia que soporta la asociación entre diferentes genotipos con serotipos y fenotipos específicos5 y probablemente en el futuro los pacientes serán clasificados por su genotipo y su perfil de autoanticuerpos lo que facilitará un mejor enfoque diagnóstico, terapéutico y pronóstico. En la actualidad se conocen varios autoanticuerpos específicos de las miopatías inflamatorias y que marcan subgrupos clínicos y pronósticos6.
Los anticuerpos anti-SRP5-7 se encuentran en alrededor del 5% de los pacientes con miopatías inflamatorias y se dirigen contra la partícula de señal de reconocimiento que es un complejo de ribonucleoproteínas (proteínas-7SLRNA) cuya función es regular la traslocación de proteínas a través del retículo endoplásmico. Los pacientes con anti-SRP cursan con miopatía necrotizante aguda y grave, con fibrosis endomisial y poco infiltrado inflamatorio, con altos niveles de CPK y pobre respuesta al tratamiento; algunos autores han encontrado asociación con miocarditis y menor frecuencia de EPID.
Los anti-Mi-25,6,8 se presentan en el 20% de los adultos y en el 4% a 10% de los niños con dermatomiositis y se dirigen contra Mi-2 que es una helicasa nuclear que juega un papel importante en la transcripción genética. Los pacientes de este subgrupo cursan con enfermedad cutánea, miopatía leve y buena respuesta al tratamiento esteroideo.
Los anticuerpos anti-P155/140 tienen como blanco T1FI γ que es factor de transcripción y diferenciación celulares; este subgrupo se caracteriza por enfermedad cutánea grave en adultos y niños y a miopatía asociada a cáncer en adultos5-10.
Anti-P140 (anti-MJ) se dirigen contra NPX-2, una proteína de la matriz nuclear comprometida en la transcripción nuclear y el metabolismo del RNA; es un marcador de dermatomiositis juvenil con calcicosis5,6,11.
El blanco antigénico de anti-SAE (Small Ubiquitin-like Modifier Activating Enzime) es un grupo de subunidades proteicas comprometidas en la modificación postraslacional; estos anticuerpos marcan un subgrupo de pacientes que inician con dermatomiositis amiopática y progresan a miositis con disfagia pero con menor frecuencia de EPID5,6,12.
Recientes estudios demuestran anticuerpos contra CADM 140, identificado como MDA-5 (cytoplasmic protein melanoma differenciation associated-gene 5), que marcan un subgrupo caracterizado por hallazgos cutáneos de dermatomiositis pero sin hallazgos significativos de miositis, aunque pueden ser subclínicos, y enfermedad pulmonar rápidamente progresiva. El hecho de que MDA-5 está comprometida en la respuesta inmune innata contra infecciones virales y que anti-CDAM 140 ha sido encontrada exclusivamente en pacientes japoneses puede sugerir que la enfermedad es iniciada por un factor ambiental (virus) en individuos genéticamente susceptibles13.
Anticuerpos antisintetasa
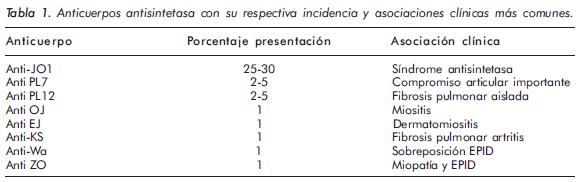
AntiJo-1 fue el primero de los anticuerpos antisintetasa caracterizado; se describió en 1976 en el suero del paciente John P (de ahí su nombre), quien debutó con un cuadro de neumonía intersticial y, de manera similar al paciente que se reporta, desarrolló miopatía inflamatoria (en su caso polimiositis) posterior al compromiso pulmonar, siendo evidente ésta una vez se inició el desmonte de esteroides. Anti-Jo-1 es el prototipo de anticuerpos cuyo blanco es la histidil-tRNA sintetasa, una enzima citoplasmática que acetila el RNA de trasferencia (Tabla 1)4. De todos los anticuerpos que componen el grupo de los antisintetasa, los anti-Jo-1 son los únicos utilizados en la práctica clínica y están presentes en 20% a 30% de los pacientes con miopatías inflamatorias4. En el contexto de síndrome antisintetasa la presencia de anticuerpos anti-Jo-1 confiere un 70% de probabilidades de tener EPID en el trascurso de la enfermedad, aunque sólo se correlaciona de manera modesta con su actividad6. Reportes de casos de anti-Jo-1 en pacientes con otras enfermedades reumáticas autoinmunes que desarrollan enfermedad pulmonar intersticial soportan el concepto de un papel patogénico directo de dicho anticuerpo en el desarrollo del síndrome antisintetasa.
Varios mecanismos explican la asociación entre la producción de dichos anticuerpos y las miopatías inflamatorias14,15. Se propone que fenómenos de mimetismo molecular con enzimas virales permiten la pérdida de inmunotolerancia16. Un ejemplo de esto es el trabajo realizado por Bowles17 en el cual se buscó virus Coxsackie en pacientes con miopatía inflamatoria, otras enfermedades musculares y controles sanos, encontrando este sólo en el tejido muscular de los pacientes afectados por algunos de los subtipos de miopatía inflamatoria. La unión del RNA viral a la histidil RNA trasferasa humana propiciaría el fenómeno de pérdida de inmunotolerancia. Una vez se producen anticuerpos anti-Jo-1, éstos inducen respuestas celulares y humorales que generan inflamación de las fibras musculares; los complejos inmunes circulantes podrían estimular macrófagos alveolares y favorecer la inflamación17. Por el contrario, la expresión enriquecida de Jo-1 en monocapas de células alveolares pulmonares clivadas con granzima B, en comparación con otros tejidos, incluido el músculo, sugiere que el tejido que inicia la respuesta autoinmune en el "síndrome antiJo-1" es el pulmón, con ataque secundario del músculo18,19.
En los últimos años se han caracterizado anticuerpos contra varios blancos antigénicos que hacen parte de los antisintetasa y que se presentan con frecuencias entre el 1% y el 5%6,7; entre ellos están: los anticuerpos anti-treonil-t RNA sintetasa (anti-PL7), anti-alanil-t RNA sintetasa (anti-PL12), anti-glicil-t RNA sintetasa (anti-EJ), anti-isoleucil -t RNA sintetasa (antiOJ), antiasparaginil -t RNA sintetasa (anti-Ks), anti-tirosil t RNA sintetasa (anti-Ha) y anti-fenilalanil -t RNA sintetasa (antiZo).
Manifestaciones clínicas
La presentación del síndrome incluye EPID, fenómeno de Raynaud, artritis y "manos de mecánico". De todas estas manifestaciones, el compromiso pulmonar es el más frecuente y el más grave, con una incidencia promedio del 70%4 y con una gran variedad de presentaciones; su inicio puede ser sincrónico, preceder o anteceder el compromiso muscular20 y su curso clínico puede ser agudo, crónico o asintomático, siendo este último evidenciado sólo en pruebas de imagen o función pulmonar20-22. Generalmente los pacientes con neumonitis intersticial presentan artralgias, síntomas generales y fiebre hasta en el 80%1 de los casos; el compromiso miopático está presente en más del 90% y el fenómeno de Raynaud entre el 30% y el 50% de ellos. El signo de las "manos de mecánico" consiste en la presencia de lesiones eritematosas hiperqueratósicas y no pruriginosas que comprometen la cara lateral y palmar de las manos, simulando una dermatitis ocupacional de los pacientes sometidos a actividad manual intensa22. Bachmeyer y colaboradores, en una serie retrospectiva de siete pacientes con síndrome antisintetasa27, describieron la asociación de este signo con el compromiso pulmonar y los anticuerpos anti-Jo-1; las manos de mecánico se presentaron en el 57% de los pacientes, siendo, en el 75% de los casos, el único signo cutáneo presente tanto en el debut como en las recurrencias del compromiso pulmonar. Si bien la mayoría de los pacientes cursan con miopatía grave y refractaria al tratamiento, el síndrome antisintetasa con EPID grave también puede observarse en pacientes con dermatomiositis amiopática24.
Diagnóstico
Los pacientes con afección respiratoria generalmente presentan, como primer síntoma, tos no productiva o disnea de esfuerzos. A la auscultación pulmonar se evidencian crépitos finos bibasales17,25. En los rayos X los hallazgos tienden a localizarse en las bases pulmonares, con la presencia de infiltrados intersticiales o mixtos pero estos cambios son tardíos, no difieren de los observados en otras causas de EPID y no son útiles para definir el compromiso pulmonar específico26. En los estudios de pacientes con síndrome antisintetasa, donde se utilizó la tomografía de alta resolución de tórax27-29, se encontró que el hallazgo más frecuente fue la consolidación subpleural, lesión reversible con el tratamiento inmunosupresor; esta fase aguda inflamatoria progresa hasta la presencia de infiltrados en panal de abeja por fibrosis pulmonar y una pobre respuesta al tratamiento. En la espirometría se observa un patrón restrictivo de la función pulmonar, generalmente precedido por una disminución de la capacidad de difusión de monóxido de carbono (DLCO), que es el método más sensible para detectar el compromiso intersticial inicial2 y tiene valor pronóstico, pues la presencia de una DLCO disminuida por debajo del 45% en la ausencia de hipertensión pulmonar es un fuerte predictor de cronicidad27,30. El hallazgo histopatológico pulmonar más común es el de neumonía intersticial no específica31.
Tratamiento
No existen ensayos clínicos controlados que evalúen medicamentos en la EPID secundaria a síndrome antisintetasa. Los esteroides tienen efectos benéficos en las manifestaciones sistémicas y en algunos subtipos de compromiso pulmonar29. En términos generales, la respuesta pulmonar es tardía con respecto a la mejoría muscular y se obtiene a expensas de altas dosis con sus subsecuentes efectos secundarios, motivo por el cual se ha descrito tratamiento adicional con: ciclofosfamida32,33, ciclosporina33, azatioprina34 y metotrexate35, en estudios observacionales. La ciclofosfamida33 se ha utilizado como terapia de inducción logrando remisión y, en ocasiones, regresión completa de los infiltrados pulmonares; la azatioprina, el metotrexate y la ciclosporina han mostrado efectividad como ahorradores de esteroides y para detener la progresión de la enfermedad a pesar de presentar recaídas con el desmonte de tratamiento30. El metrotexate, por su posible toxicidad pulmonar, es generalmente evitado. El tacrolimus se puede considerar como una alternativa en casos refractarios37. La inmunoglobulina intravenosa ha sido estudiada para el manejo de miopatía inflamatoria sin tener un papel claro en el tratamiento de la EPID. Späth39 y colaboradores describieron su experiencia con 12 pacientes con síndrome antisintetasa, encontrando en dos de ellos una respuesta adecuada; no obstante, debe recordarse el carácter transitorio del efecto de las inmunoglobulinas, pudiendo tener algún papel en el control inicial del cuadro. Franzonili y colaboradores40 proponen seleccionar los inmunosupresores con base en las subpoblaciones de células inflamatorias predominantes en el lavado broncoalveolar (BAL)41; en su estudio, al inicio de la enfermedad pulmonar intersticial, seleccionaron los pacientes con diagnóstico de síndrome antisintetasa y compromiso pulmonar dado por evidencia de fibrosis pulmonar o disminución de la capacidad de difusión de CO2 por debajo del 80% y los dividieron en dos grupos: los pacientes con predominio de linfocitos eran asignados a tratamiento combinado con azatioprina y colchicina y los pacientes con predominio de neutrófilos o eosinófilos a manejo con ciclofosfamida; la terapia esteroidea era reservada solo para controlar las manifestaciones musculares a dosis bajas requiriendo en promedio 5 mg/día de prednisona o su equivalente. En el seguimiento a 24 meses el compromiso pulmonar permaneció estable en todos los pacientes. Este trabajo tiene limitaciones importantes como el número reducido de pacientes o la presencia exclusiva de mujeres en la muestra, pero sugiere un esquema de tratamiento basado en la celularidad del BAL y deja planteados varios interrogantes, entre ellos la real pertinencia del manejo con esteroides en este tipo de pacientes y el potencial uso de colchicina con sus propiedades antifibróticas.
Pronóstico
El compromiso pulmonar es el principal determinante de la sobrevida de los pacientes con síndrome antisintetasa24; esto se demuestra en una revisión de la literatura1 que incluye 62 pacientes y demuestra una mortalidad a 32 meses de 40%, siendo al menos la mitad de las causas secundarias al compromiso pulmonar de la enfermedad. Sin embargo, la mayoría de estos pacientes sólo recibieron esteroides y sólo cinco de ellos recibieron algún inmunosupresor adicionado tarde en el curso de su enfermedad. En un estudio de la universidad de Kyoto42 que revisa el pronóstico de 140 pacientes con miopatía inflamatoria, se encontró enfermedad intersticial pulmonar en el 50%; esta frecuencia es mayor que la reportada en otras series (30%)18, indicando una diferencia étnica que realza la importancia de contar con series locales. En esta cohorte, el 32% de los pacientes debutaron con enfermedad pulmonar intersticial y posteriormente presentaron la miopatía inflamatoria; estos pacientes tuvieron mayor asociación con anticuerpos antisintetasa y mayor tendencia a las recaídas. Es importante recalcar que este subgrupo de pacientes pueden diagnosticarse como artritis reumatoide (AR) por la alta frecuencia de sinovitis (57%) y de factor reumatoide (30%)43. En el estudio mencionado un tercio de los pacientes recibieron manejo con DMARDS por un diagnóstico inicial de AR.
Los anticuerpos antisintetasa no son los únicos asociados a EPID en los pacientes con miopatías inflamatorias; la presencia de anti-Ro se correlacionó con una mayor severidad radiológica y en la DLCO4.
La presencia de neutrófilos en el BAL y la presentación de un cuadro clínico similar al síndrome de Hamman-Rich se acompaña de un pronóstico pobre al corto plazo, con refractariedad al tratamiento y progresión a fibrosis pulmonar31. La bronquiolitis obliterante con neumonía organizada generalmente tiene buena respuesta a esteroides, siendo quizás el único subtipo de la enfermedad en el cual se observa una respuesta importante a estos medicamentos. La presencia de imágenes en panal de abejas indica la progresión de la enfermedad con fibrosis asociada y deterioro de la función pulmonar en el mediano a largo plazo31.
En conclusión, las miopatías inflamatorias autoinmunes corresponden a un amplio grupo de entidades con diferentes autoanticuerpos, los cuales son marcadores de compromiso orgánico y de subgrupos clínicos y pronósticos; los anticuerpos antisintetasa marcan un síndrome clínico grave y cuya primera manifestación puede ser la EPID como en el caso informado.
Referencias
1. Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet 2003;362(9388):971-982. [ Links ]
2. Amato AA, Barohn RJ. Inclusion body myositis: old and new concepts. J Neurol Neurosurg Psychiatry 2009;80(11):1186-1193. [ Links ]
3. Dalakas MC. Polymyositis, Dermatomyositis and Inclusion Body Myositis. In: Fauci AS, Braunwald E, Kasper DL, Hauser SL, Longo DL, Jameson JL, et al, editors. Harrison's Principles of Internal Medicine. 17 ed. New York: Mc Graw Hill 2007;2696-2703. [ Links ]
4. Imbert-Masseau A, Hamidou M, Agard C, GRolleau JY, Chérin P. Antisynthetase Syndrome. Joint Bone Spine. 2003;70(3):161-168. [ Links ]
5. Sato S, Hirataka M, Kuwana M. Autoantibodies to a 140 KD polypeptides, CDAM 140, in japanese patient with clinically amyipathic dermatomyositis. Arth Rheum 2005;52:1571-1576. [ Links ]
6. Targoff IN. Autoantibodies and their significance in myositis. Current Rheumatology Rep 2008; 10:333-340. [ Links ]
7. Gunawardena H, Betteridge ZE, McHugh N. Myositisspecific autoantibodies: their clinical and pathogenic significance in disease expression. Rheumatology 2009; 48: 607-612 . [ Links ]
8. Hengstman GJ, ter L Wang HB, Zhang Y. Mi2, an autoantigen for Dermatomyositis is an ATP-depending nucleosoma remodelling factor. Nucleic Acid Res 2001;29:2517-2521. [ Links ]
9. Wang HB, Zhang Y. Mi2, an autoantigen for Dermatomyositis is an ATP-depending nucleosoma remodelling factor. Nucleic Acid Res 2001;29:2517-2521. [ Links ]
10. Gunawardena H, North J, Weddernburn L. Clinical association of antiP155/140 in adult and juvenile Dermatomyositis (Abstract). Ann Rheum Dis 2007; 66:S68. [ Links ]
11. Chinoy H, Fertig N, Addis CV, Ollier WE, Cooper RD. The diagnostic utility of myositis autoantibodies testing for predicting the risk of cancer associated myositis. Ann Rheum Dis 2007;66:1345-1349. [ Links ]
12. Targoff IN, Trieu EP, Levy-Neto M, Fertig N, Oddis CV. Sera with autoantibodies to the MJ antigen reacts with NPX-2. Arthritis Rheum 2007;56:S787. [ Links ]
13. Levine SM, Raven N, Xie D. Novel conformation of histidyl-RNA transfer syntetase in the lung; a target tissue in Jo-1-associated myositis. Arth Rheum 2007;56:2729-2739. [ Links ]
14. Stone KB, Oddis CV, Ferting N, Katsumata Y, Lucas M, Vogt M, et al. AntiJo-1 antibody levels correlate with disease activity in idiopathic inflammatory myophaty. Arthritis Rheum 2007;56(9):3125-3131. [ Links ]
15. Ascherman DP, Oriss TB, Oddis CV, Wright TM. Critical requirement for professional APCs in eliciting T cell responses to novel fragments of histidyl-tRNA synthetase (Jo-1) in Jo-1 antibody-positive polymyositis. J Immunol 2002;169(12):7127-7134. [ Links ]
16. Mimori T, Imura Y, Nakashima R, Yoshifuji H. Autoantibodies in idiopathic inflammatory myopathy: an update on clinical and pathophysiological significance. Curr Opin Rheumatol 2007;19(6):523-529. [ Links ]
17. Bowles NE, Dubowitz V, Sewry CA, Archard LC. Dermatomyositis, polymyositis, and Coxsackie-B-virus infection. Lancet 1987;1(8540):1004-1007. [ Links ]
18. Fathi M, Lundberg IE. Interstitial lung disease in polymyositis and dermatomyositis. Curr Opin Rheumatol 2005;17(6):701-706. [ Links ]
19. Levine SM, Raven N, Xie D. Novel conformation of histidyl-RNA transfer syntetase in the lung; a target tissue in Jo-1-associated myositis. Arth Rheum 2007;56:2729-2739. [ Links ]
20. Bowles NE, Dubowitz V, Sewry CA, Archard LC. Dermatomyositis, polymyositis, and Coxsackie-B-virus infection. Lancet 1987;1(8540):1004-1007. [ Links ]
21. Hirakata M, Nagai S. Interstitial lung disease in polymyositis and dermatomyositis. Curr Opin Rheumatol 2000; 12(6):501-508. [ Links ]
22. Frazier AR, Miller RD. Interstitial pneumonitis in association with polymyositis and dermatomyositis. Chest 1974;65(4):403-407. [ Links ]
23. Hirakata M, Nagai S. Interstitial lung disease in polymyositis and dermatomyositis. Curr Opin Rheumatol 2000; 12(6):501-508. [ Links ]
24. Pinto L F, Velásquez CJ, Márquez JD. Síndrome antisintetasa en un paciente con dermatomiositis amiopática. Rev Col Reumatol 2008;15(4):331. [ Links ]
25. AlJanadi M, Smith CD, Karsh J. Cyclophosphamide treatment of interstitial pulmonary fibrosis in polymyositis/ dermatomyositis. J Rheumatol 1989;16(12):1592-1596. [ Links ]
26. Costabel U, King TE . International consensus statement on idiopathic pulmonary fibrosis. Eur Respir J 2001;17(2):163-167. [ Links ]
27. Bachmeyer C, Tillie-Leblond I, Lacert A, Cadranel J, Aractingi S. 'Mechanic's hands': a misleading cutaneous sign of the antisynthetase syndrome. Br J Dermatol 2007;156(1):192-194. [ Links ]
28. Marie I, Hachulla E, Chérin P, Dominique S, Hatron PY, Hellot M F, et al. Interstitial lung disease in polymyositis and dermatomyositis. Arthritis Rheum 2002;47(6):614-622. [ Links ]
29. Bonnefoy O, Ferretti G, Calaque O, Coulomb M, Bequeret H, Beylot-Barry M. Serial chest CT findings in interstitial lung disease associated with polymyositisdermatomyositis. Eur J Radiol 2004;49(3):235-244. [ Links ]
30. Douglas WW, Tazelaar HD, Hecker PA , Schroeder DR, et al. Polymyositis-dermatomyositis-associated interstitial lung disease. Am J Respir Crit Care Med 2001;164(7):1182-1185. [ Links ]
31. Tansey D, Wells AU, Colby TV, Ip S, Nikolakopolou A , du Bois RM, et al. Variations in histological patterns of interstitial pneumonia between connective tissue disorders and their relationship to prognosis. Histopathology 2004;44(6):585-596. [ Links ]
32. AlJanadi M, Smith CD, Karsh J. Cyclophosphamide treatment of interstitial pulmonary fibrosis in polymyositis/ dermatomyositis. J Rheumatol. 1989;16(12):1592-1596. [ Links ]
33. Schnabel A, Reuter M, Gross WL. Intravenous pulse cyclophosphamide in the treatment of interstitial lung disease due to collagen vascular disease. Arth Rheum 1998;41(7):1215-1220. [ Links ]
34. Qushmaq KA, Chalmers A, Esdaile JM. Cyclosporine A in the treatment of refractory adult polymyositis/ dermatomyositis: population based experience in 6 patients and literature review. J Rheumatol 2000;27(12):2855-2859. [ Links ]
35. Bunch TW, Worthington JW, Combs JJ, Ilstrup DM, Engel AG. Azathioprine and prednisone for polymyositis: a controlled clinical trial. Ann Intern Med 1980;92(3):365-369. [ Links ]
36. Joffe MM, Love LA, Leff RL, Fraser DD, Targoff IN, Hicks JE, et al. Drug therapy of the idiopathic inflammatory myopathies: predictors of response to prednisone, azathioprine, and methotrexate and a comparison of their efficacy. Am J Med 1993;94:379-387. [ Links ]
37. Wilkes MR, Sereika SM, Fertig N, Lucas MR, Oddis CV. Treatment of Antisynthetase-Associated Interstitial Lung Disease With Tacrolimus. Arthritis Rheum 2005;52(8):2439-2446. [ Links ]
38. Douglas WW, Tazelaar HD, Hecker PA, Schroeder DR, et al. Polymyositis-dermatomyositis-associated interstitial lung disease. Am J Respir Crit Care Med 2001;164(7):1182-1185. [ Links ]
39. Späth M, Schröder M, Sholotter-Weigel B, Walter MC, Hautmann H, Leinsinger G, et al. The long-term outcome of antiJo-1-positive inflammatory myopathies. J Neurol 2004;251:859-864. [ Links ]
40. Franzolini N, Quartuccio L, De Marchi G, De Vita S. Efficacy of ab initio immunosuppressive therapy and steroid sparing effect in interstitial lung disease associated to antisynthetase syndrome. Reumatismo 2007;59(3):202-208. [ Links ]
41. Sauty A, Rochat T, Schoch OD, Hamacher J, Kurt AM, Dayer JM. Pulmonary fibrosis with predominant CD8 lymphocytic alveolitis and antiJo-1 antibodies. Eur Respir J 1997;10(12):2907-2912. [ Links ]
42. Yoshifuji H, Fuji T, Kobayashi S, Imura Y, Fujita Y, Kawabata D, et al. Anti-aminoacyl-tRNA synthetase antibodies in clinical course prediction of interstitial lung disease complicated with idiopathic inflammatory myopathies. Autoimmunity 2006;39(3):233-241. [ Links ]
43. Hirakata M, Nagai S. Interstitial lung disease in polymyositis and dermatomyositis. Curr Opin Rheumatol. 2000;12(6):501-508. [ Links ]

