










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Reumatología
Print version ISSN 0121-8123
Rev.Colomb.Reumatol. vol.18 no.1 Bogotá Jan./Mar. 2011
PRESENTACIÓN DE DOS CASOS Y REVISIÓN DE LA LITERATURA
2Médico Internista Reumatólogo, Universidad de Antioquia.
3Médico Internista Reumatólogo, Hospital Pablo Tobón Uribe. Medellín, Antioquia.
4Médico Internista Reumatólogo, Hospital San José, Bogotá.
Correspondencia: Luis Fernando Pinto lfpintop@hotmail.com
Los autores declaran no presentar ningún conflicto de interés al momento de la redacción del manuscrito.
Recibido: 18 de diciembre de 2010. Aceptado: 3 de febrero de 2011
Resumen
Informamos dos pacientes clasificadas como síndrome antifosfolipídico primario que presentaron hemorragia alveolar y nefropatía durante su evolución. La primera paciente presentó una glomerulonefritis membranoproliferativa con patrón "full house" en la inmunofluorescencia; los autoanticuerpos específicos de LES fueron persistentemente negativos y los niveles de complemento sérico permanecieron normales; la segunda paciente presentó una proteinuria en rango no nefrótico y síndrome HELLP y, posteriormente, una hemorragia pulmonar. Ninguna de las pacientes ha desarrollado un cuadro definitivo de LES luego de varios años de seguimiento.
Palabras clave: hemorragia alveolar, síndrome antifosfolipídico, nefropatía.
Summary
We report two patient with primary antiphospholipid syndrome with pulmonary haemorraghe and nefropathy. First patient had a membranoprolipherative glomerulonephritis and shown "full house pattern" in her renal biopsy; specific SLE autoantibodies were negative and complement levels was normal. The second patient presents with no nephrotic proteinuria, HELLP syndrome followed by pulmonary hemorrhage. They don't developed full blown SLE during several years of follow up.
Key words: alveolar hemorrhage, antiphospholipid syndrome, nephropathy.
Introducción
La hemorragia alveolar difusa (HAD) es una condición amenazante de la vida que puede ser causada por un grupo diverso de enfermedades; generalmente se comporta como una emergencia médica que frecuentemente ocasiona falla respiratoria aguda, por lo cual requiere de un rápido diagnóstico y un tratamiento intensivo1. Esta entidad puede ser la expresión clínica de una capilaritis pulmonar. Dentro de las posibles causas de HAD se encuentran: las vasculitis asociadas a ANCAs, varias enfermedades autoinmunes, trastornos de la coagulación, medicamentos y condiciones diversas; algunos casos permanecen sin causa definida. Dentro de este grupo de entidades sobresalen el síndrome de Behçet, el síndrome de Goodpasture, la poliangiitis microscópica, el lupus eritematoso sistémico (LES), el síndrome antifosfolipídico y la granulomatosis de Wegener2.
La HAD se caracteriza clínicamente por disnea y hemoptisis, infiltrados alveolares difusos bilaterales en la radiografía del tórax y anemia; debe ser distinguida de otras causas de acumulación de eritrocitos en el espacio alveolar. En la HAD verdadera, el lavado broncoalveolar a menudo permite observar fibrina intraalveolar y hemosiderina en las paredes alveolares así como hemosiderófagos1.
El síndrome antifosfolipídico (SAF) es una enfermedad autoinmune caracterizada por eventos trombóticos o complicaciones gestacionales recurrentes en presencia de anticuerpos antifosfolipídicos. Inicialmente descrito en el contexto de LES, posteriormente se reconoció también como una enfermedad independiente (SAF primario) o asociada con otras enfermedades autoinmunes3.
Caso 1
Mujer de 42 años con diagnóstico previo de SAF primario por presentar anticoagulante lúpico (LA) positivo y una historia obstétrica consistente en seis gestaciones, dos hijos vivos, tres abortos entre las semanas 8 y 12 y una pérdida fetal a las 24 semanas. Cinco años antes había presentado un episodio de depresión mayor con características psicóticas y desarrolló epilepsia del tipo de crisis focal compleja, sin eventos trombóticos previos o citopenias hematológicas; la paciente venía en tratamiento con cloroquina 150 mg/día y ácido valproico 750 mg/día; y tenía antecedente de alergia al ácido acetilsalicílico.
A nuestro hospital fue remitida por riesgo de falla respiratoria tras un cuadro de tres días de evolución consistente en astenia, adinamia, disfonía, disnea progresiva de esfuerzos hasta el reposo, tos, expectoración hemoptoica y fiebre subjetiva. Negaba síntomas gastrointestinales, urinarios, dolor torácico, úlceras orales, caída del cabello, fotosensibilidad y artritis. Manifestaba haber tenido úlceras en el tabique nasal y en los pies.
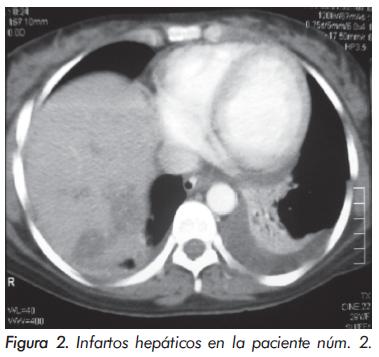
Al examen físico se encontró en buenas condiciones, con dificultad respiratoria, pálida y disfónica. Frecuencia cardiaca: 96 por minuto, presión arterial: 123/78, frecuencia respiratoria: 17 por minuto, saturación de oxígeno: 97% (recibiendo oxígeno por cánula nasal), afebril. Sus mucosas estaban húmedas, sin lesiones orales, con placas eritematosas en el tabique nasal del lado izquierdo, sin perforación. No presentaba dilatación yugular ni masas en cuello. Los ruidos cardiacos eran regulares, con soplo sistólico mitral grado II/VI, sin frote ni galope, y se encontró disminución global de ruidos respiratorios, con escasos crépitos en ambas bases. Extremidades sin edemas ni asimetrías, sin déficit de pulsos, sin cianosis distal, con úlcera en quinto dedo de pie izquierdo y en la cabeza del quinto metatarsiano del pie derecho. Al examen neurológico se encontró alerta, orientada, sin focalización motora, encefalopatía ni signos de irritación meníngea. Se observaba fenómeno de Raynaud en los miembros superiores. Los resultados de laboratorio se consignan en la Tabla 1.
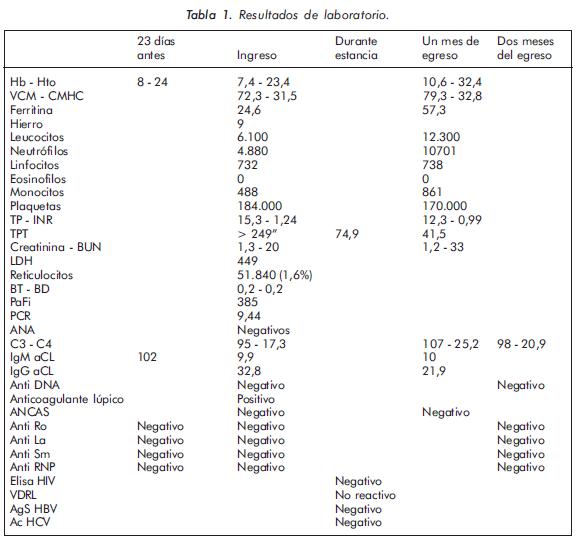
La radiografía de tórax mostró ocupación alveolar difusa (Figura 1) y la tomografía axial computadorizada de tórax de alta resolución imágenes con patrón en vidrio esmerilado.
Tres días después de su ingreso se le realizó fibrobroncoscopia con lavado bronco-alveolar (BAL), se tomaron muestras para microbiología incluyendo cultivos para micobacterias y hongos que fueron negativos; en la citología del BAL predominaron los macrófagos y los neutrófilos. Se observa ron algunas células del epitelio respiratorio y neumocitos con cambios reactivos. La coloración de hierro fue positiva con presencia de abundantes hemosiderófagos. El conjunto de la presentación clínica y radiológica, la hemoglobina baja a su ingreso y los hallazgos en el lavado broncoalveolar confirman el diagnóstico de hemorragia alveolar. La tomografía de cráneo demostró la presencia de varios infartos corticales antiguos.
Al ingreso se utilizaron antimicrobianos de amplio espectro los cuales fueron rápidamente suspendidos al descartar infección. Posteriormente se realizó tratamiento con metilprednisolona 1 gramo diario por cinco días y ciclofosfamida 1 gramo dosis única con resolución de los síntomas respiratorios y de los infiltrados pulmonares luego de siete días.
Durante la primera semana de hospitalización la paciente presentó proteinuria de 600 mg en 24 horas, creatinina 1,2 mg/dL, nitrógeno ureico 54 mg/dL y sedimento urinario activo; la biopsia renal mostró una glomerulonefritis membranoproliferativa con "patrón full house" en la inmunofluorescencia (depósitos de IgG, IgM, IgA, C3, C1q y cadenas livianas Kappa y Lambda) por lo que se ha estudiado con ANAs, antiDNA y antiENAs que han mostrado resultados negativos y complemento sérico que ha sido normal en varias ocasiones durante tres años de seguimiento. Se trató con prednisolona 1 mg/kg/día en dosis decrecientes y ciclofosfamida 1 gramo IV mensual por seis meses, logrando una remisión completa de la glomerulonefritis (proteinuria menor de 500 mg/día, sedimento urinario limpio y función renal normal); la terapia de mantenimiento se hizo con micofenolato 2 gr/día. El tratamiento actual de la paciente consiste en micofenolato 1,5 gramos/día y clopidogel 75 mg/día y se encuentra en remisión completa; los exámenes de su última cita, tres años después del episodio de HAD, muestran anticuerpos AntiDNA y antiENAs negativos, C3 y C4 normales, hemoglobina de 12,3 gramos/dL, 6500 leucocitos, 4300 neutrófilos y 1900 linfocitos y 187000 plaquetas/ mm3.
Caso 2
Mujer de 29 años de edad con diagnóstico de SAF primario de cinco años de evolución por haber presentado cuatro abortos durante el primer trimestre, títulos altos de anticardiolipinas IgG e IgM y anticoagulante lúpico fuertemente positivo; nunca había presentado trombosis o citopenias hematológicas y venía en tratamiento con 100 mg/día de aspirina. Aunque la paciente presentó anticuerpos antinucleares (ANA) positivos 1:1280 con patrón moteado fino y títulos bajos de anticuerpos anti-Ro en una ocasión, estos fueron posteriormente negativos; nunca tuvo criterios clínicos de LES y los anticuerpos anti-DNA fueron persistentemente negativos.
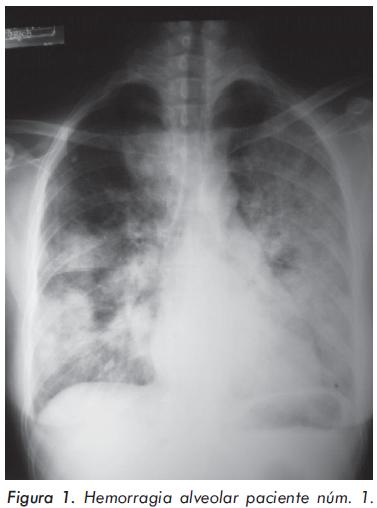
Ingresó al servicio de urgencias con un embarazo de 21 semanas, por presentar edema de miembros inferiores, cifras tensionales elevadas, y dolor y distensión abdominal de una semana de evolución. Los estudios iniciales mostraron proteinuria en rango no nefrótico, hematuria, anemia hemolítica, 36.000 plaquetas /mm3 y elevación de las transaminasas cuatro veces por encima del rango superior normal. Se decidió interrumpir el embarazo por preeclampsia severa y síndrome HELLP y se trató con cinco pulsos de metilprednisolona (1 gramo/día) y 1 gramo de ciclofosfamida endovenosa; sin embargo, no hubo mejoría de la hemólisis y la trombocitopenia, por lo cual se le administró gamaglobulina endovenosa; se documentaron varios infartos hepáticos por tomografía abdominal (Figura 2) e infección de tejidos blandos por staphilococcus aureus que fue tratada con antibióticos. Cuatro días después la paciente presentó deterioro de su estado general y falla respiratoria que requirió soporte ventilatorio; se documentó hemorragia alveolar por lo que se agregaron recambios plasmáticos al manejo con buena respuesta. Después de salir de la unidad de cuidados intensivos, la paciente presentó pancitopenia que fue tratada con Rituximab 500 mg/semana por cuatro dosis y pulsos de metilprednisolona y ciclofosfamida con mejoría transitoria pero, un mes después, presentó enfermedad cerebrovascular trombótica del territorio de la arteria cerebral posterior y recidiva de la pancitopenia. Durante la hospitalización no se documentaron anticuerpos antiDNA positivos ni hipocomplementemia.
Discusión
Los pacientes con SAF pueden desarrollar un espectro amplio de enfermedad pulmonar. El compromiso pulmonar en el SAF es frecuente e incluye embolismo e infarto pulmonar, hipertensión pulmonar tromboembólica, hipertensión arterial pulmonar, síndrome de dificultad respiratoria del adulto, hemorragia intraalveolar y capilaritis pulmonar4.
Aunque el estado hipercoagulable es el mecanismo patogénico más común en la afectación pulmonar por el SAF, existe un número creciente de reportes que describen patología pulmonar en ausencia de trombosis. Dentro de estos se reportan hipertensión pulmonar no tromboembólica, vasculopatía pulmonar y hemorragia alveolar debida a capilaritis pulmonar. La HAD con capilaritis pulmonar es una complicación rara del SAF, con sólo 20 casos publicados entre 1966 y 20065.
Se ha propuesto que en el SAF primario la HAD es debida a capilaritis pulmonar inducida por acción directa de los anticuerpos antifosfolípidos. Es posible, sin embargo, que coexista algún grado de trombosis junto con el proceso de vasculitis predominante. Los anticuerpos antifosfolípidos inducen un aumento en la expresión de moléculas de adhesión en las células endoteliales con el consecuente reclutamiento y migración de neutrófilos hacia los septos alveolares, generando destrucción tisular y hemorragia. Adicionalmente la activación del complemento inducida por anticuerpos antifosfolipídicos puede llevar también a activación de los neutrófilos mediada por la fracción C5a contribuyendo con este proceso inflamatorio6.
La HAD es una situación potencialmente fatal y podría ser la manifestación inicial de SAF; sin embargo, en el contexto de esta entidad, la HAD es un diagnóstico de exclusión y deberán descartarse siempre otras enfermedades autoinmunes sistémicas como LES, granulomatosis de Wegener y poliangiitis microscópica y otras causas como edema pulmonar, uremia, coagulopatía, émbolos pulmonares e infección6,7.
La mayoría de los pacientes que son tratados con pulsos de glucocorticoides mejoran, independiente de si se adiciona o no ciclofosfamida. Por lo tanto el tratamiento de primera línea de la HAD en SAF son los glucocorticoides (usualmente dosis altas endovenosas). Ante la alta posibilidad de recurrencia se propone usar otros inmunosupresores como ciclofosfamida, ciclosporina o micofenolato. Si existe sangrado activo podría ser necesario interrumpir la anticoagulación en caso que el paciente la reciba hasta que el estado pulmonar haya mejorado, y reiniciarla en caso de estar indicada con mucha precaución. En casos refractarios al manejo convencional se han usado la inmunoglobulina endovenosa y los recambios plasmáticos6.
La afectación renal en los pacientes con SAF es infrecuente, menor del 10%, y se manifiesta principalmente por microangiopatía trombótica8; también se han descrito hiperplasia fibrosa de la íntima, atrofia cortical focal y oclusiones arteriales9 presentándose como hipertensión arterial, proteinuria en rango no nefrótico e insuficiencia renal y estenosis de las arterias renales10. Hasta en el 32% de las biopsias renales de los pacientes con nefritis lúpica pueden encontrarse lesiones compatibles con SAF renal agregadas a los cambios inflamatorios11. Los hallazgos histológicos sugestivos de SAF renal son: trombos de fibrina en las arteriolas y los glomérulos, proliferación de la intima por miofibroblastos, trombos organizados con recanalización de la luz y retracción subcapsular del parénquima renal con atrofia y fibrosis.
Más controvertida es la presentación de glomerulonefritis en pacientes con SAF primario porque en la mayoría de los casos publicados se presenta asociación con ANAs, antiDNA y consumo de complemento lo que sugiere LES; Sinico y cols.12 encuentran afectación renal en el 8,7% de 160 pacientes con SAF primario; en diez biopsias renales demostraron cuatro casos de glomerulonefritis membranosa, dos con glomerulonefritis proliferativa difusa, dos con microangiopatía trombótica y dos con lesiones vasculares compatibles con SAF crónico. Las biopsias que demostraron glomerulonefritis membranosa presentaron depósitos inmunes subepiteliales y mesangiales y presencia de C3 y de C1q; dos de estos pacientes cursaron con títulos bajos de ANAs e hipocomplementemia. Los dos pacientes con glomerulonefritis proliferativa difusa presentaron depósitos inmunes mesangiales y subepiteliales, proliferación mesangial y "patrón full house" en la inmunofluorescencia y cursaron con hipocomplementemia y títulos bajos de ANAs y antiDNA.
A diferencia de los casos descritos por Sinico y cols., nuestra primera paciente ha tenido persistentemente negativos los autoanticuerpos y normales los niveles séricos de complemento, cumple criterios para SAF y no tiene datos clínicos ni serológicos concluyentes de LES u otras enfermedades autoinmunes, por lo cual consideramos que se trata de un caso de SAF primario; sin embargo, la presentación de "patrón full house" en la inmunofluorescencia, en especial el depósito de C1q, obliga a seguirla para detectar la presencia de un LES12.
En los últimos años se le ha dado una creciente importancia al papel del complemento en la mediación de la trombosis y de las pérdidas fetales13; la presentación de glomerulonefritis con depósitos de complemento puede ser la demostración del papel de los mediadores inflamatorios en otras manifestaciones no obstétricas ni trombóticas del SAF y no necesariamente corresponder a la asociación con LES.
En nuestra segunda paciente se demostraron ANAs y títulos bajos de anticuerpos antiRo al inicio de su enfermedad; sin embargo, ha desarrollado criterios para LES durante cinco años de seguimiento; varios autores14,15, informan la presencia de títulos bajos de ANAs, anticuerpos antiDNA y antiENAs en pacientes con SAF primario; en nuestro grupo el 25% de los pacientes con SAF primario cursaron con ANAs positivos y el 10% con anticuerpos antiDNA o antiENAs en títulos bajos16.
En nuestras dos pacientes no realizamos biopsia pulmonar para demostrar capilaritis pulmonar por la gravedad de su estado, pero el cuadro clínico, los hallazgos radiológicos, la anemia, la demostración de hemosiderófagos en el lavado broncoalveolar y la exclusión de infección y otros diagnósticos diferenciales, nos permiten considerar que las pacientes desarrollaron hemorragia alveolar difusa asociada con síndrome antifosfolipídico primario; sin embargo, hasta el 10% de los pacientes clasificados como SAF primario pueden evolucionar a otras enfermedades autoinmunes, en especial a LES, luego de varios años de la primera manifestación trombótica u obstétrica17. Nuestras pacientes son un ejemplo más de las fronteras poco definidas que pueden existir entre las diferentes enfermedades autoinmunes, en este caso el SAF primario y el LES18.
Conclusión
La HAD es una complicación poco frecuente pero potencialmente fatal del SAF primario. Los clínicos deben mantener un alto índice de sospecha de esta complicación en un paciente con síntomas respiratorios agudos, anemia progresiva e infiltrados pulmonares. Frecuentemente es necesario usar antimicrobianos empíricos mientras se descarta la posibilidad de infección pulmonar aislada o asociada; sin embargo, se debe tener precaución en no atribuir los síntomas pulmonares a un proceso infeccioso sin una evaluación detallada, lo que retardaría el tratamiento inmunosupresor con consecuencias potencialmente fatales. La glomerulonefritis proliferativa difusa con "patrón full house" en la inmunofluorescencia es una manifestación rara del SAF primario; este caso realza la importancia de los diferentes mecanismos inflamatorios implicados en la inmunopatogénesis del SAF. Presentaciones clínicas atípicas del SAF, como la hemorragia pulmonar y la glomerulonefritis, obligan a vigilar estrechamente el desarrollo de un LES.
Referencias
1. Collard HR, Schwarz MI. Difusse alveolar hemorrhage. Clin Chest Med, 2004;25:583-592. [ Links ]
2. Ioachimescu OC, Stoller JK. Diffuse alveolar hemorrhage: diagnosing it and finding the cause. Clev Clin J Med 2008;75(4):258-280. [ Links ]
3. Grossman JM. Primary versus secondary antiphospholipid syndrome: is this lupus or not? Curr Rheum Rep 2004;6:445-450. [ Links ]
4. Espinosa G, Cervera R, Font J, Asherson RA. The lung in the antiphospholipid syndrome. Ann Rheum Dis 2002;61:195-198. [ Links ]
5. Marcos Rodríguez PJ, Montero Martínez C, Verea Hernando H. Hemorragia pulmonar y síndrome antifosfolípido primario: aportación de un caso y revisión de la literatura. An Med Interna 2007;24(3):125-128. [ Links ]
6. Deane KD, West SG. Antiphospholipid antibodies as a cause of pulmonary capillaritis and difuse alveolar hemorrhage: a case series and literature review. Sem Arthritis Rheum 2005;35:154-165. [ Links ]
7. Green RJ, Ruoss SJ, Kraft SA, Raffin TA, Berry GJ. Pulmonary capillaritis and alveolar hemorrhage: update on diagnosis and Management. Chest 1996;110;1305-1316. [ Links ]
8. Amigo MC, García-Torres R, Robles M, Bochicchio T, Reyes PA. Renal involvement in primary antiphospholipid síndrome. J Rheumatol 1992;9:181-185. [ Links ]
9. Tektonidou MG, Sotsiou F, Nakopoulou L, Vlachoyiannopoulos PG, Moutsopoulos HM. Antiphospholipid syndrome nephropathy in patients with systemic lupus erythematosus and antiphosphospholipid antibodies: prevalence, clinical associations and long term outcome. Artrhritis Rheum 2004;50:2569-2579. [ Links ]
10. Paul SN, Sangle SR, Benett AN. Vasculitis, antiphospholipid antibodies, and renal artery stenosis. Ann Rheum Dis 2005;64:1800-1802. [ Links ]
11. Daugas E, Nochy D, Huong D, Dehaut P, Beauflis H, Caudwell B, et al. Antiphospholipid Syndrome Nephropathy in Systemic Lupus Erythematosus. J Am Soc Nephrol 2002;13:42-52. [ Links ]
12. Sinico RA, Cavazana I, Nuzzo M, Vianelli M, Napodano P, Scaini P, et al. Renal involvement in primary antiphospholipid syndrome: Retrospective analysis of 160 Patients. Clin J Am Soc Nephrol 2010;5:1211-1217. [ Links ]
13. Pierangeli SS, Pojen P, Chen PP, Raschi E, Scurati S, Grossi C, et al. Antiphospholipid antibodies and the Antiphospholipid Syndrome: pathogenic mechanisms. Sem Throm Hemost 2008;34:236-250. [ Links ]
14. Vianna JL, Khamashta MA, Ordi-Ros J, Font J, Cervera R, Lopez-Soto A, et al. Comparison of the primary and secondary antiphospholipid syndrome. A European multicentric study of 131 patients. Am J Med 1994;96:3-9. [ Links ]
15. Asherson MA, Khamashta MA, Ordi-Ros J, et al. The "primary" antiphospholiíd syndrome: mayor clinical and serological features. Medicine 1989;68:366-374. [ Links ]
16. Vargas F, Pinto LF, Molina JF, Donado J, Eraso R, Tobón JA, et al. Síndrome Antifosfolípido: Morbilidad y evolución de una cohorte de pacientes del Hospital Pablo Tobón Uribe de Medellín-Colombia. Rev Col Reum 2006;13:109-119. [ Links ]
17. Gómez-Puerta JA, Martín H, Amigo MC, Aguirre MA, Camps MT, Cuadrado MJ, et al. Long-term follow-up in 128 patients with primary antiphospholipid syndrome: do they develop lupus? Medicine 2005;84:225-230. [ Links ]
18. Shoenfeld Y, Moroni PL, Toubi E. Antiphospholipid Syndrome and Systemic lupus erhytematosus: are they separate entibies or just clinical presentations on the same scale? Curr Op Rheumatol 2009;21:495-500. [ Links ]

