










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Reumatología
Print version ISSN 0121-8123
Rev.Colomb.Reumatol. vol.18 no.2 Bogotá Apr./june 2011
PRESENTACIÓN DE CASO Y REVISIÓN DE LA LITERATURA/CASE REPORT AND LITERATURA REVIEW
1Residentes Reumatología, Universidad de Antioquia.
2Profesor, Departamento de Patología, Facultad de Medicina, Universidad de Antioquia.
3Profesor, Departamento de Medicina Interna, Sección de Reumatología, Facultad de Medicina, Universidad de Antioquia.
Correspondencia: Luis Alonso González. Correo electrónico: lagnvvn@hotmail.com
Los autores declaran no presentar ningún conflicto de interés al momento de la redacción del manuscrito. No hubo ningún tipo de financiación.
Recibido: 07 de marzo de 2011. Aceptado: 24 de abril de 2011
Resumen
Informamos el caso de un hombre de 79 años con hemorragia alveolar, glomerulonefritis necrosante y polineuropatía por poliangiítis microscópica, una enfermedad autoinmune, caracterizada por vasculitis sistémica, predominantemente de vasos de pequeño calibre. Se revisan los aspectos clínicos y terapéuticos de la enfermedad.
Palabras clave: poliangiítis microscópica, autoanticuerpos contra el citoplasma de neutrófilos (ANCA), hemorragia alveolar, glomerulonefritis necrosante.
Summary
We report the case of a 79 years-old male, with alveolar hemorrhage, necrotizing glomerulonephritis and polyneuropathy due to microscopic polyangiitis, an autoimmune disease, characterized by systemic vasculitis, predominantly of small vessels. We review the clinical and therapeutic aspects of the disease.
Key words: microscopic polyangiitis, antineutrophil cytoplasmic autoantibodies (ANCA), alveolar hemorrhage, necrotizing glomerulonephritis.
Introducción
La poliangiítis microscópica (PAM) es una vasculitis sistémica que afecta principalmente a vasos pequeños, aunque también, puede afectar vasos de mediano calibre. Característicamente, se asocia con la presencia de anticuerpos contra el citoplasma de los neutrófilos (ANCA), por lo que es clasificada como una vasculitis asociada a ANCA, un subgrupo de vasculitis primarias en la que también se incluyen la granulomatosis de Wegener (GW), el síndrome de Churg-Strauss (SCS) y la vasculitis limitada al riñón. Este tipo de vasculitis puede manifestarse clínicamente como un síndrome pulmón-riñón con una glomerulonefritis rápidamente progresiva y hemorragia alveolar, sin embargo sus manifestaciones clínicas varían según el órgano afectado1. Presentamos el caso de un paciente con poliangiítis microscópica que se manifestó con insuficiencia renal, hemorragia alveolar y polineuropatía sensitivo-motora, y realizamos una revisión de la literatura de esta forma de vasculitis sistémica.
Caso clínico
Hombre de 79 años, con antecedente de hipertensión arterial diagnosticada dos años atrás y ex tabaquismo. Consultó por síntomas de 4 meses de evolución consistentes en expectoración hemoptoica intermitente, pérdida de 6 kilogramos de peso, astenia, deterioro en la clase funcional hasta la postración, mialgias y disminución de la fuerza muscular en miembros inferiores y superiores. Al examen físico su presión arterial era de 160/100 mmHg, y en la auscultación cardiopulmonar se encontraron crépitos en ambas bases pulmonares; la fuerza muscular se encontró disminuida tanto proximal (4/5) como distal (2/5) en miembros superiores e inferiores, con hiporreflexia y flacidez. Como consecuencia de ello, presentaba caída bilateral de manos y pies (figura 1).
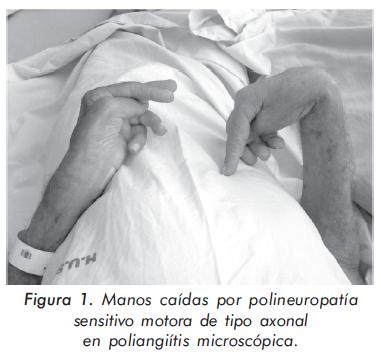
Los exámenes de laboratorio de ingreso mostraron: anemia normocítica normocrómica (Hemoglobina 10,5 g/dL, VCM de 88 fL), velocidad de sedimentación globular de 120 mm/hora, proteína C reactiva de 20 mg/dL, ferritina sérica de 1010 ng/ml, creatinina sérica de 1,5 mg/dL, disminución en la depuración de creatinina (16 ml/min) y proteinuria en rango no nefrótico (900 mg/24 horas). En el sedimento urinario se encontró piuria (50 leucocitos por campo de alto poder) y hematuria microscópica (8-12 eritrocitos por campo de alto poder). En la radiografía de tórax se encontraron infiltrados intersticiales bibasales, y en la ecocardiografía se documentó hipertrofia del ventrículo izquierdo con fracción de eyección del 80%. La electromiografía y estudio de conducción nerviosa de cuatro extremidades demostró la presencia de una polineuropatía sensitivo motora de tipo axonal.
Al quinto día de estancia hospitalaria presentó fiebre (38,8°C), disnea y expectoración hemoptoica. La radiografía de tórax, esta vez, mostró un incremento en los infiltrados pulmonares comprometiendo los cuatro cuadrantes y la tomografía de tórax de alta resolución fue interpretada como una neumonía multilobar. Se detectaron dos hemocultivos positivos para Klebsiella pneumoniae, por lo cual se inició tratamiento con piperacilina tazobactam. Aunque hubo resolución de la fiebre y se negativizaron los hemocultivos, los infiltrados pulmonares en los estudios radiológicos progresaron y los reactantes de fase aguda permanecieron elevados (proteína C reactiva de 24 mg/dl). Al realizar la fibrobroncoscopia se encontró una endobronquitis moderada en el árbol bronquial derecho, sin sangrado; no se aislaron microorganismos en las tinciones directas ni en los cultivos. Sin embargo, la presencia de hemosiderófagos de > 50% en el lavado broncoalveolar fue compatible con el diagnóstico de hemorragia alveolar.
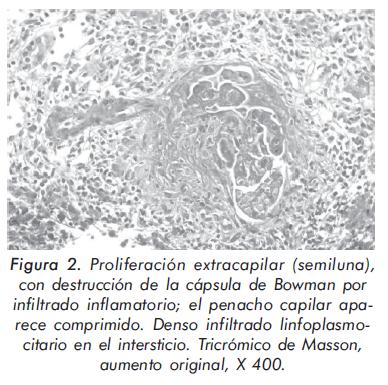
Los anticuerpos contra el citoplasma de los neutrófilos (ANCA) fueron positivos (1:160) con patrón periférico (p-ANCA) en la inmunofluorescencia indirecta con especificidad para mieloperoxidasa (anti-MPO) en la prueba de ELISA (74,9 UI). Los hallazgos histológicos de la biopsia renal demostraron una glomerulonefritis necrosante focal y segmentaria con vasculitis (figura 2), mientras que en la biopsia de nervio sural se observó reacción inflamatoria en el perineuro, necrosis fibrinoide en los vasos y células inflamatorias en el interior de algunas fibras nerviosas en el endoneuro. Con estos hallazgos, se hizo el diagnóstico de PAM.
Se inició tratamiento con 50 mg/día de prednisolona y pulsos intravenosos de ciclofosfamida (CYC), obteniéndose rápida mejoría de la afección pulmonar, estabilización de la polineuropatía y mejoría en las pruebas de función renal (creatinina 1,3 mg/dl y depuración de creatinina de 34 ml/min). En su última evaluación, luego de recibir dos pulsos de CYC (dosis acumulada de 1,5 gramos) y prednisolona (50 mg/día) se logró la resolución de los infiltrados alveolares, y las pruebas de función renal mostraron una creatinina sérica de 1,2 mg/dl y una depuración de creatinina en 33 ml/min.
Poliangiítis microscópica
Descrita por primera vez en 1923 por Friedrich Wohlwill2 y posteriormente revisada por Davson y cols. en 19483, la PAM fue clasificada en la Conferencia de Consenso de Chapel Hill (CCCH), en 1994, como una vasculitis necrosante de vasos pequeños caracterizada por la usencia de depósitos de complejos inmunes en la inmunofluorescencia y la presencia de capilaritis pulmonar y glomerulonefritis. Asimismo, se establecieron diferencias con la poliarteritis nodosa (PAN) clásica, definida como una vasculitis necrosante de vasos medianos sin glomerulonefritis o vasculitis en arteriolas, capilares o vénulas (tabla 1)4.

Asociación con los anticuerpos contra el citoplasma de los neutrófilos (ANCA)
Los anticuerpos dirigidos contra los constituyentes de los gránulos azurófilos de los neutrófilos y los monocitos fueron descritos por primera vez por Davies y cols. en 19825. Posteriormente, estos anticuerpos fueron relacionados con la GW6 y más tarde con la PAM7. En la técnica de inmunofluorescencia indirecta, con neutrófilos humanos fijados en etanol como sustrato, estos anticuerpos exhiben dos patrones de inmunofluorescencia: un patrón citoplasmático (cANCA) en el que se produce una tinción granular del citoplasma, y un patrón perinuclear (pANCA) en el que la tinción es periférica o difusa del núcleo. El patrón de inmunofluorescencia de los ANCA suele correlacionarse, en mayor o menor grado, con el tipo de vasculitis en el que aparecen. Así, en la GW, el patrón mayoritariamente detectado es el cANCA y en la PAM es el pANCA8. También los pacientes afectados de SCS y vasculitis limitada a riñón suelen exhibir un patrón perinuclear. Respecto a la especificidad de estos autoanticuerpos, se han identificado mediante la prueba de ELISA (derivado del inglés, enzime-linked immunoassays), dos tipos principales de autoanticuerpos, uno que reacciona con la proteinasa-3 (PR3ANCA) y que produce el patrón citoplasmático en la IFI9 y otro que reacciona con la mieloperoxidasa (MPO-ANCA) y que produce el patrón perinuclear (pANCA) en la IFI7.
Aunque algunos autores sugieren que los MPO-ANCA y los PR3-ANCA se asocian con el diagnóstico de PAM con una frecuencia casi igual10, el grupo de Chapel Hill encontró un predominio de los MPO-ANCA en esta forma de vasculitis11.
Epidemiología
La PAM tiene un ligero predominio en hombres (relación hombre: mujer de 1,5-1,8:1) y una edad promedio de inicio entre los 50 y los 60 años12,13. El primer estudio que sugiere un incremento en la incidencia de la PAM proviene de Leicester, Reino Unido, donde la incidencia anual de PAM entre 1980 y 1985 fue de 0,5 por millón de habitantes y entre 1987 y 1989 de 3,3 por millón de habitantes14. Estudios más recientes sugieren una mayor incidencia durante los 1990s que los 1980s15. En Europa, se ha informado una variación geográfica en la proporción de la incidencia de la GW : PAM, siendo mayor la incidencia de PAM en el sur de Europa y mayor la incidencia de GW en el norte de Europa16. Es probable que tal incremento en la incidencia de la PAM sea por las definiciones establecidas en la CCCH donde se establecen sus diferencias con la PAN clásica y la GW y por un mejor conocimiento de esta enfermedad por parte de los médicos (tabla 1).
Varios estudios epidemiológicos han tratado de dilucidar los factores ambientales asociados con el inicio de las vasculitis y algunos autores han encontrado asociaciones con la exposición a sílice y a solventes17.
Patogénesis
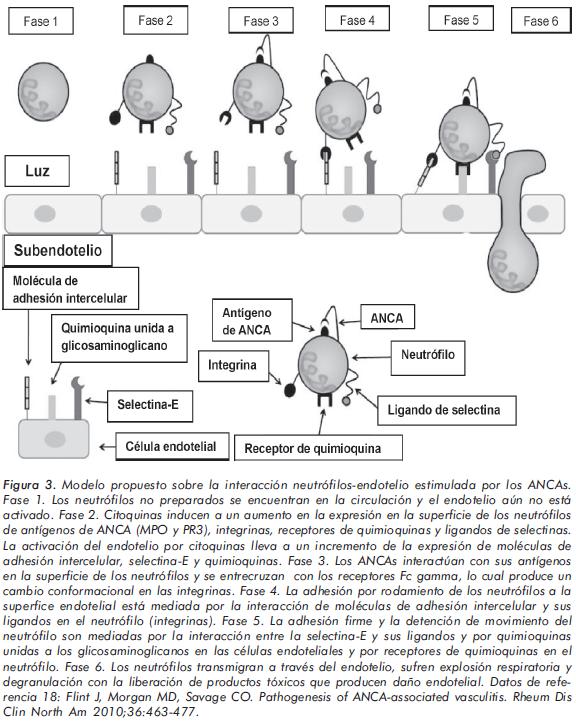
La evidencia actual posiciona a los ANCA como elementos importantes en la patogénesis de la PAM. Estos autoanticuerpos (MPO-ANCA) tienen la capacidad de activar a los neutrófilos, que bajo estímulo de diferentes citoquinas (interleuquina-1 y factor de necrosis tumoral-a), han expresado en su supeficie mieloperoxidasa (MPO) y se han adherido al endotelio vascular (figura 3)12,18. El papel de los MPO-ANCA en la patogénesis de la PAM se ha demostrado en modelos animales, en los cuales, estos anticuerpos inducen el desarrollo de capilaritis pulmonar y glomerulonefritis19. Sin embargo, no todos los pacientes con PAM y vasculitis activa son ANCA positivos y los títulos de estos autoanticuerpos se correlacionan pobremente con la actividad de la enfermedad. Estas observaciones indican que los ANCA no son esenciales en la patogénesis en todos los pacientes con PAM y que probablemente existe más de un mecanismo implicado en la génesis de esta vasculitis18, 20.
Manifestaciones clínicas
A diferencia de la GW, la cual usualmente se manifiesta con signos y síntomas característicos como sinusitis, la PAM puede manifestarse insidiosamente con signos inespecíficos de inflamación. De hecho, más del 70% los pacientes tienen síntomas constitucionales al momento del diagnóstico, principalmente pérdida de peso y fiebre. El cuadro clínico puede tener una presentación aguda en días o semanas o un curso indolente a lo largo de meses o años antes del diagnóstico. Como consecuencia de ello, en el momento del diagnóstico, los órganos, especialmente los riñones, pueden tener una enfermedad avanzada con un deterioro importante en su función12,21.
Afección renal. Es el principal hallazgo clínico de la PAM y se caracteriza por una glomerulonefritis rápidamente progresiva; sin embargo, puede variar desde un sedimento urinario patológico asintomático hasta una enfermedad renal terminal que requiere diálisis. Entre el 80% y el 100% de los pacientes presentan manifestaciones renales sin importar la especificidad de los ANCA22,23. Las manifestaciones clínicas más frecuentes de la afección renal son la proteinuria (en rango nefrótico hasta en el 50% de los pacientes), hematuria microscópica y cilindros eritrocitarios y granulares en el sedimento urinario23. La hipertensión arterial se encuentra en un 25% de los pacientes con PAM21.
El hallazgo característico en la biopsia renal es una glomerulonefritis necrosante focal y segmentaria, que se observa hasta en el 100% de los pacientes con afección renal. Las semilunas glomerulares también son frecuentes y se observan en aproximadamente el 90% de los pacientes. Una franca vasculitis con necrosis fibrinoide es menos frecuente, estando presente sólo en el 20% de los estudios histológicos renales. La nefritis intersticial y la atrofia tubular se encuentran en la mitad de los pacientes con PAM23.
En la inmunofluorescencia hay ausencia o escasos depósitos de inmunoglobulinas o complemento en el glomérulo y los vasos renales, por consiguiente, el término pauci-inmune. Dado que los hallazgos observados en la histología renal son similares en los tres tipos de vasculitis asociadas a ANCA (PAM, GW, SCS), ésta no es útil para diferenciarlas. Sin embargo, en biopsias renales de pacientes con vasculitis asociada con MPO-ANCA se observa un patrón de mayor cronicidad de la enfermedad como fibrosis intersticial, atrofia de los túbulos y glomeruloesclerosis24. Estas diferencias pueden ser debido a un retraso en el diagnóstico en los pacientes con PAM en comparación con los pacientes con GW.
Los anticuerpos ANCA y los anticuerpos antimembrana basal glomerular (anti-MBG) pueden coexistir. El 30% de los pacientes con anticuerpos anti-MBG tienen ANCA, mientras que el 5% a 14% de los pacientes con ANCA positivos tienen antiMBG. En estos pacientes, la mayoría de los ANCA están dirigidos contra la MPO (66%-100%)25.
Afección pulmonar. Las manifestaciones pulmonares están presentes en el 25% a 55% de los pacientes e incluyen: hemorragia alveolar, derrame pleural, fibrosis intersticial, edema pulmonar, pleuritis y, con menos frecuencia, panbronquiolitis y aneurisma de la arteria pulmonar. De éstas, la manifestación pulmonar clásica, es la hemorragia alveolar difusa causada por capilaritis, la cual ha sido informada en el 12% a 55% de los pacientes; los pacientes con hemorragia alveolar tienen una mayor tasa de recaídas y de mortalidad26,27.
Aunque puede estar limitada a los pulmones, en un estudio retrospectivo de 29 pacientes con PAM y hemorragia alveolar, la afección pulmonar se asoció con al menos un órgano extrapulmonar afectado, siendo el riñón el más comprometido, en el 97% de esos pacientes27.
En las radiografías de tórax se observan opacidades con un patrón de llenado alveolar, con mayor frecuencia bilateral y en campos pulmonares superiores e inferiores. La afección exclusiva de los campos pulmonares superiores se ha descrito en el 30% de estos pacientes21. El hallazgo más frecuente en la tomografía computarizada es la atenuación en vidrio esmerilado (> 90% de los pacientes), lo cual corresponde a hemorragia alveolar, inflamación crónica intersticial de los septos alveolares y capilaritis. Las consolidaciones se observan en el 80% de los pacientes con afección pulmonar. También se puede apreciar imágenes en panal de abejas12.
En cuanto a las pruebas de laboratorio, un descenso súbito en los valores de hemoglobina de más de 1 g/dl en 48 horas, es sugestivo de hemorragia alveolar. Los ANCA, dos tercios con un patrón perinuclear y un tercio con un patrón citoplasmático, se encuentran en más del 90% de los pacientes con enfermedad pulmonar grave. El líquido del lavado broncoalveolar es usualmente hemorrágico23. La tinción con azul de Prusia de Perls de la muestra del lavado broncoalveolar muestra un elevado número de macrófagos cargados de hemosiderina, los cuales se encuentran en más del 30% de los pacientes con PAM26,27. Aunque rara vez se realiza, la lesión histológica observada en la biopsia pulmonar es una capilaritis necrosante pauciinmune y el sangrado alveolar. La presencia de granulomas sugiere como diagnostico una GW.
Las pruebas de función pulmonar pueden mostrar un patrón restrictivo o un patrón obstructivo. La capacidad de difusión de monóxido de carbono usualmente está disminuida, sin embargo, ésta puede incrementarse durante una hemorragia alveolar activa12.
La fibrosis pulmonar es otro hallazgo cada vez más reconocido de la PAM. Puede preceder por meses o años, aparecer al momento o después del diagnóstico. Esta manifestación ha sido asociada con episodios previos de hemorragia alveolar y tiene un mal pronóstico10,21.
Afección cutánea. Las manifestaciones cutáneas se encuentran en el 30% a 60% de los pacientes y al momento del diagnóstico pueden estar presentes en el 15% a 30%. La manifestación más frecuente es la púrpura palpable, presente en un 30% a 40%28. Otros hallazgos cutáneos informados han sido livedo reticularis, nódulos, urticaria y úlceras cutáneas con necrosis29.
En la histología de biopsias obtenidas de la púrpura palpable se observa una vasculitis leucocitoclástica con infiltrado neutrofílico de vasos de pequeño calibre localizados en la dermis superficial, necrosis fibrinoide y polvo nuclear. En algunos casos se ha informado la presencia de infiltrado linfocitario perivascular. La biopsia de nódulos subcutáneos muestra afección de los vasos de la dermis profunda29. La inmunofluorescencia puede ser negativa, o mostrar, escasos depósitos de inmunoglobulinas o complemento. Aunque los nódulos son más frecuentes en la PAN y la púrpura palpable es más frecuente en la PAM, ambos hallazgos pueden presentarse en esas dos enfermedades. Por tanto, estas manifestaciones y sus hallazgos histológicos no permiten diferenciar entre PAN y PAM12.
Afección neurológica. Las manifestaciones neurológicas se presentan hasta en un 72% de los pacientes. La neuropatía periférica es más frecuente que la afección del sistema nervioso central y las presentaciones más frecuentes son la mononeuritis múltiple y la polineuropatía simétrica distal. Los nervios afectados en orden de frecuencia descendente son: peroneo superficial, peroneo profundo, cubital, mediano y, muy raramente, los nervios craneales21. En estudios de conducción nerviosa se detecta una axonopatía, mientras que en los hallazgos histológicos se demuestra una vasculitis necrosante, que se observa en biopsias de nervio sural hasta en el 80% de los pacientes afectados30. La documentación histológica de isquemia del nervio puede ser suficiente para hacer el diagnóstico de vasculitis, incluso en ausencia de necrosis fibrinoide de la arteria epineural21.
Las manifestaciones del sistema nervioso central representan hasta un 30% de la afección neurológica de la PAM. Algunas de estas manifestaciones son: hemorragia cerebral, paquimeningitis e infartos cerebrales no hemorrágicos31,32.
Afección musculoesquelética. Al momento del diagnóstico, el 56% al 76% de los pacientes tienen mialgias y artralgias. Los niveles séricos de creatina fosfoquinasa y de aldolasa son normales, al igual que la resonancia magnética. Al igual que en otras vasculitis necrosantes, la artritis es rara y cuando se presenta, no es destructiva21.
Afección gastrointestinal. El dolor abdominal, la diarrea y el sangrado intestinal ocurren en el 30% a 58% de los pacientes, siendo el dolor abdominal el más frecuente. Aunque el sangrado abdominal se presenta en el 20% a 30% de los pacientes, la hemorragia masiva es inusual. Mediante la angiografía, se ha demostrado la presencia de aneurismas arteriales como la causa del sangrado. Otras manifestaciones gastrointestinales menos frecuentes son: úlceras del colon, isquemia y perforación intestinal33.
La enfermedad hepática es rara. Los hallazgos histológicos muestran degeneración fibrinoide de las arterias interlobulares y arteritis necrosante e infiltrado linfocitario de los tractos portales12,34.
Afección cardiaca. La afección de vasos pequeños del corazón es bastante rara. En una cohorte de 85 pacientes franceses el 17,6% presentaron falla cardíaca y el 10% pericarditis22.
Afección ocular. Manifestaciones como epiescleritis, escleritis, iridociclitis y vasculitis de la retina han sido observadas durante el curso de la enfermedad. La epiescleritis se ha informado hasta en el 20% de los pacientes10.
Otras manifestaciones. Aunque no se considera como una manifestación clínica de la PAM, sino más bien de la GW o del SCS, la afección de oídos, nariz y garganta se ha informado en un 9%, 20% y 30% de los pacientes respectivamente23. La sinusitis se ha informado hasta en un 11% de los pacientes22. Estas observaciones probablemente se deban a una superposición entre la PAM y la GW, pero también podrían indicar que son manifestaciones poco usuales de la PAM a las cuales no se les ha hecho mucha énfasis21.
Diagnóstico diferencial
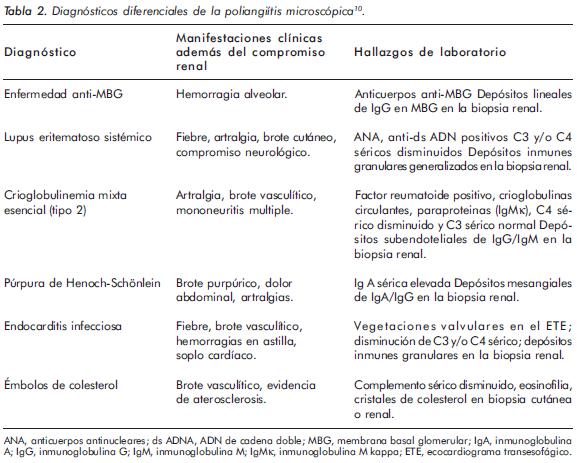
Debido a que los síntomas iniciales como fiebre, mialgias y artralgias son inespecíficos, los pacientes usualmente llegan al especialista con alteraciones en la función renal, y en algunos casos, con hemorragia alveolar (síndrome pulmón-riñón). Hay varios diagnósticos diferenciales a tener en cuenta los cuales se muestran en la tabla 210.
Pruebas de laboratorio
No se cuenta en la actualidad con ninguna prueba de laboratorio específica para establecer el diagnóstico de PAM. Dado que los ANCA están presentes el 50-75% de los pacientes, su ausencia no excluye el diagnóstico de PAM. Los ANCA asociados con PAM generalmente exhiben un patrón de inmunofluorescencia perinuclear (pANCA) el cual es causado por anticuerpos antiMPO, detectados por la técnica de ELISA. En una cohorte de pacientes franceses con PAM, los ANCA fueron positivos en 38 de 51 pacientes (74,5%) y de ellos 33 (86,8%) tuvieron un patrón p-ANCA y 5 un patrón c-ANCA. Los anticuerpos anti-MPO fueron detectados por ELISA en 31 pacientes y los anticuerpos anti-PR3 en 4 pacientes22.
La técnica de inmunofluorescencia indirecta tiene una mayor sensibilidad y la prueba de ELISA una mayor especificidad para el diagnóstico de PAM. Sin embargo, estos autoanticuerpos pueden estar presentes en otras vasculitis asociadas a ANCA, en otras enfermedades inflamatorias y en algunas infecciones12. Por tanto, para establecer el diagnóstico se requiere la integración del cuadro clínico, la histología y los autoanticuerpos.
Cuando la prueba de inmunofluorescencia muestra un patrón perinuclear, pero la prueba de ELISA es negativa, el resultado de la prueba de ANCA no se puede considerar positiva ya que ambas pruebas deben ser positivas para asegurar la especificidad diagnóstica21.
Otras pruebas de laboratorio como las pruebas de función renal, parcial de orina, medición de proteínas en orina deben evaluarse rutinariamente durante el seguimiento de los pacientes. En cuanto a los marcadores de inflamación, los hallazgos más comunes son la elevación de la velocidad de sedimentación globular y la proteína C reactiva. También puede haber leucocitosis, trombocitosis y anemia normocítica normocrómica35.
Pronóstico
Aunque la PAM es considerada una enfermedad grave, también se han descrito formas leves22 y, en general, su pronóstico ha mejorado. Debido a que los pacientes afectados por PAM son de mayor edad (edad promedio de 61 años) que los pacientes con diagnóstico de GW y SCS (53 años y 50 años en promedio, respectivamente), su tasa de mortalidad es más alta (27,5%) que la de la GW (13,2%) y la del SCS (13,9%)36. Las recaídas en la PAM son frecuentes, pero menos que en la GW 37 y por lo general responden al tratamiento.
El "Puntaje de Cinco Factores" [en inglés, FiveFactor Score (FFS)], una herramienta diseñada para evaluar el pronóstico al momento del diagnóstico de la PAN, el SCS y la PAM en una cohorte francesa de 342 pacientes, es utilizada por los clínicos para evaluar la gravedad de estas vasculitis necrosantes. El FFS también se emplea en algunas circunstancias para tomar decisiones en la elección de la primera línea de tratamiento y, más concretamente, para determinar la necesidad de inmunosupresores cuando los factores de mal pronóstico están presentes. En el FFS se incluyeron los parámetros asociados con un mayor riesgo de mortalidad: proteinuria > 1 g/día, insuficiencia renal [creatinina sérica > 1,58 mg/dL (140 μmol/L)], cardiomiopatía, manifestaciones gastrointestinales graves (sangrado, perforación, infartos o pancreatitis), y afección del sistema nervioso central. Las tasas mortalidad a 5 años para un FFS de 0 (ausencia de los 5 factores), 1 (sólo un factor presente) y 2 (2 ó más factores presentes) fueron del 12%, 26% y 46%, respectivamente38.
Recientemente, se publicó una nueva versión del FFS, en la que se incluyeron pacientes con las mismas vasculitis necrosantes (PAN, SCS y PAM) y, además, con GW, no incluida en la versión inicial. Con un número mayor de pacientes (n=1108), los factores predictivos de mal pronóstico fueron: insuficiencia renal [creatinina sérica > 1,7 mg/dL (150 μmol/L)], insuficiencia cardíaca, afección gastrointestinal, edad > 65 años y ausencia de manifestaciones en oídos, nariz, y garganta, este último factor aplicable únicamente en pacientes con SCS y GW. Según esta versión, las tasas de mortalidad a 5 años para un FFS de 0,1 y > 2 fueron del 9%, 21% y 40%, respectivamente36.
Tratamiento
Este tipo de vasculitis necrosante se trata de forma similar a la GW, por diversas razones: en primer lugar, en la mayoría de los estudios prospectivos diseñados para tratar la GW se han incluido también pacientes con PAM, debido a sus características en común; en segundo lugar, no se han realizado estudios que incluyan exclusivamente pacientes con PAM; y en tercer lugar, para la mayoría de los autores, el tratamiento de la PAM no difiere del utilizado en las otras vasculitis asociadas a ANCA21. En general en PAM, el tratamiento a instaurar dependerá de la gravedad de la afección, así, para situaciones amenazantes de la vida o afección grave de órganos como glomerulonefritis, hemorragia alveolar o FFS ≥ 1, la terapia será más intensa con glucocorticoides (GCs) e inmunosupresores, a diferencia de los casos menos graves con función renal preservada en los que la monoterapia con GCs sería el tratamiento de primera línea. Debido a que la mayoría de los pacientes con PAM tienen glomerulonefritis, la monoterapia con GCs se prescribe en menos de un 20% de los pacientes que no presentan factores de mal pronóstico21.
Los GCs e inmunosupresores, especialmente la ciclofosfamida (CYC), representan la primera línea de tratamiento en la fase de inducción de la remisión en la mayoría de los pacientes. El estudio controlado de distribución aleatoria, realizado por el EUVAS (del inglés, European Vasculitis Study group), demostró que el tratamiento de inducción de la remisión con pulsos endovenosos de CYC (15 mg/kg cada 2 a 3 semanas hasta inducir remisión y luego por otros 3 meses) es igual de efectivo a la administración oral diaria de CYC (2 mg/kg/día hasta inducir remisión y luego 1,5 mg/kg/día por 3 meses más)39. Sin embargo, a largo plazo, la administración de pulsos de CYC en intervalos prolongados, no logra mantener la remisión de la enfermedad40. Por lo tanto, una vez lograda la remisión se debe instaurar una terapia de mantenimiento más constante y con menor potencial tóxico21.
El metotrexate (MTX) no ha sido evaluado como terapia de inducción en PAM, pero si como terapia de inducción para GW sin compromiso renal. A corto plazo es tan eficaz como la CYC, pero a largo plazo es menos efectivo y las recaídas son más frecuentes. Debido a su toxicidad tubular, no se recomienda en pacientes con insuficiencia renal. No obstante, el MTX es una alternativa como tratamiento de inducción para el grupo de pacientes con PAM sin afección renal21,41.
Para la terapia de mantenimiento se utiliza azatioprina (2 mg/kg/día) o MTX (0,3 mg/kg/semana inicialmente, con un incremento de 2,5 mg/ semana hasta una dosis de 25 mg/semana) que como se demostró en el estudio WEGENT (del inglés, Wegener's Granulomatosis-Entretien), tienen una eficacia y una tasa de recaídas similares37. Leflunomida (20 a 30 mg/día) puede ser más efectivo que el MTX como terapia de mantenimiento, pero se asocia con más efectos adversos20. Por su parte, el micofenolato mofetil (MMF), en un estudio de reciente publicación por el grupo EUVAS, fue menos efectivo que la azatioprina para mantener la remisión; por tanto, el MMF no está indicado como terapia de mantenimiento de primera línea42.
Los estudios clínicos conducidos por el EUVAS han demostrado que en las vasculitis asociadas a ANCA, la respuesta a los diferentes protocolos de manejo depende de la gravedad de la enfermedad41,43. Por ello, han propuesto categorizar la vasculitis en: localizada si hay afección de tracto respiratorio superior o inferior, sin síntomas constitucionales ni afección sistémica; sistémica temprana para cualquier tipo de manifestación que no represente peligro para el órgano afectado o la vida; generalizada si hay insuficiencia renal con creatinina < 5,6 mg/ dl (500 μmol/L) u otra manifestación que represente riesgo para otro órgano afectado; grave si hay falla renal con creatinina > 5,6 mg/dl o falla de otro órgano vital; y refractaria para la enfermedad progresiva que no responde a GCs o a CYC20.
Esta categorización ha sido incluida en las recomendaciones de manejo del EULAR (del inglés, European League Against Rheumatism) de 2009 (tabla 3), en las que se indica para inducción de la remisión en vasculitis de asociadas a ANCA: uso de metotrexate oral o parenteral (15 mg/semana inicialmente, hasta 20-25 mg/semana) y GCs para la vasculitis sistémica temprana; CYC (2 mg/kg/día vía oral, ó 15 mg/kg IV cada 15 días los 3 primeros pulsos, y luego cada 3 semanas hasta completar 6 a 9 pulsos) y GCs (1 mg/ Kg/día de prednisolona o su equivalente) para la enfermedad generalizada; y en vasculitis grave CYC, GCs y plasmaféresis44.
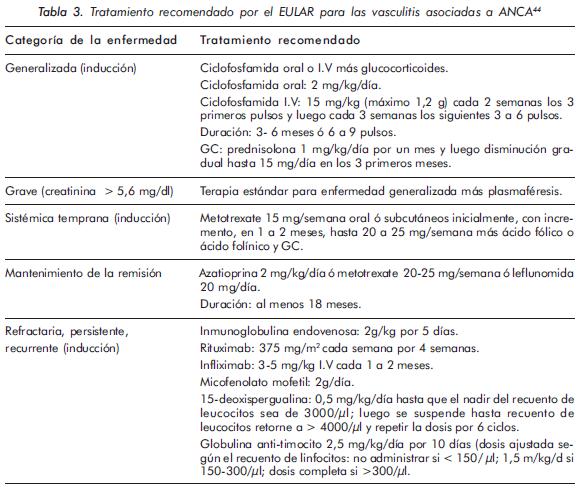
Respecto a la plasmaféresis, el estudio realizado por Jayne y colaboradores que incluyó 137 pacientes (42 con diagnóstico de GW y 95 con diagnóstico de PAM), comparó la adición de plasmaféresis con la adición de pulsos de metilprednisolona a la terapia con CYC y prednisona en vasculitis renal grave (creatinina > 5,8 mg/dl), y confirmó que la adición de plasmaféresis fue significativamente superior a los pulsos de metilpredniolona en la mejoría de la función renal, aunque no impactó significativamente la sobrevida, lo cual respalda su uso en PAM cuando la afección es grave45.
Otras terapias como la inmunoglobulina IV (IgIV), los inhibidores del factor de necrosis tumoral alfa (anti TNF-α) y los anticuerpos Anti CD20, también han sido estudiadas en vasculitis asociadas a ANCA, teniendo indicaciones específicas. La IgIV (2 g/kg en 5 días) es un medicamento seguro y bien tolerado, que interfiere con la red idiotipo anti-idiotipo, y ha sido evaluada para recaídas en diferentes estudios, demostrando una remisión completa en el 59% de los pacientes46-48. Por consiguiente, la IgIV, podía utilizarse junto con GC e inmunosupresores en pacientes que presentan recaídas durante la terapia inmunosupresora o poco después de terminada ésta. Sin embargo, dado que su principal contraindicación es la insuficiencia renal y no se recomienda para pacientes con una tasa de filtración glomerular < 30 ml/min, su prescripción en PAM está restringida a un subgrupo de pacientes21. Los anti TNF-α han sido estudiados en casos refractarios al tratamiento inmunosupresor de vasculitis asociadas a ANCA, principalmente en GW; el uso de infliximab (3 a 5 mg/kg cada 1 a 2 meses) sólo está indicado en casos refractarios de difícil manejo, por un corto periodo de tiempo (pocas semanas o meses) y no como terapia de mantenimiento, teniendo siempre presente que estos medicamentos pueden favorecer la presencia de vasculitis49. Respecto a rituximab, dos estudios recientes: el primero de ellos, el RITUXVAS (del inglés, Rituximab versus Cyclophosphamide in ANCA-Associated Vasculitis) que comparó el régimen rituximab (375 mg/m2 de superficie corporal cada semana, por cuatro dosis) más GC y CYC (15 mg/kg endovenosos con la primera y tercera infusión de rituximab) frente a CYC endovenosa (por 3 a 6 meses) más GC, para inducción de la remisión en vasculitis renal asociada a ANCA, en 44 pacientes; en este estudio el régimen basado en rituximab no fue superior a la terapia convencional con CYC y las tasas de remisión fueron altas con ambos regímenes. En el estudio RAVE (del inglés, Rituximab in ANCAAssociated Vasculitis), un estudio de no inferioridad en el que comparó rituximab (375 mg/m2 de superficie corporal cada semana, por cuatro dosis) con CYC (2 mg/kg/día), y que incluyó 197 pacientes con vasculitis asociada a ANCA (48 de ellos con PAM), demostró que la terapia con rituximab no es inferior al tratamiento diario con CYC para inducción de la remisión en vasculitis grave asociada a ANCA; además, también se observó una mayor efectividad con régimen basado en rituximab para inducción de remisión en recaídas50-51; por el momento está indicado en casos refractarios y recaídas.
Referencias
1. Scott DGI, Watts R. Polyarteritis nodosa and microscopic polyangiitis. In: Hochberg MC, Silman AJ, Smolen JS, Weinblatt, Weisman MH, eds. Rheumatology. 4th ed. Philadelphia: Elsevier 2008;1511-1519. [ Links ]
2. Wohlwill F. Uber die nur Mikroskopich erkenbarre From der Periarteritis nodosa. Arch Pathol Anat 1923;246:377-411. [ Links ]
3. Davson J, Ball J, Plat R. The kidney in periarteritis nodosa. QJ Med 1948;67:175-202. [ Links ]
4. Janette J, Falk RJ, Andrassy K, Bacon PA, Churg J, Gross WL, et al. Nomenclature of systemic vasculitis. Proposal of an international consensus conference. Arthrirtis Rheum 1994;37:187-192. [ Links ]
5. Davies DJ, Moran JE, Nial JF, Ryan GB. Segmental necrotising glomerulonephritis with antineutrophil antibody: posible arbovirus aetiology? Br Med J 1982;285:606. [ Links ]
6. van der Woude FJ, Rasmussen N, Lobatto S, Wiik A, Permin H, van Es LA, et al. Autoantibodies against neutrophils and monocytes tool for diagnosis and marker of disease activity in Wegener's granulomatosis. Lancet 1985;1:425-429. [ Links ]
7. Falk RJ, Janette JC. Anti-neutrophil cytoplasmic autoantibodies with specificity for myeloperoxidase in patients with systemic vasculitis and idiopathic necrotizing and crescentic glomerulonephritis. N Engl J Med 1988;318:1651-1657. [ Links ]
8. Jennette JC, Falk RJ. Anti-neutrophil cytoplasmic autoantibodies and associated diseases: a review. Am J Kidney Dis 1990;15:517-529. [ Links ]
9. Goldschmeding R, Dolman KM, van den Ende ME, van der Meer-Gerritsen CH, Sonnenberg A, dem Borne AE. The relation of 29 kD C-ANCA antigen to proteinase 3. APMIS Suppl 1990;19:26-27. [ Links ]
10. Smyth L, Gaskin G, Pusey CD. Microscopic polyangiitis. Semin Respir Crit Care Med 2004;25:523-533. [ Links ]
11. Falk RJ, Nachman PH, Hogan SL, Jennette JC. ANCA glomerulonephritis and vasculitis: a Chapel Hill perspective. Semin Nephrol 2000;20:233-243. [ Links ]
12. Chung SA, Seo P. Microscopic polyangiitis. Rheum Dis Clin North Am 2010;36:545-558. [ Links ]
13. Ntatsaki E, Watts RA, Scott DG. Epidemiology of ANCA-associated vasculitis. Rheum Dis Clin North Am 2010;36:447-461. [ Links ]
14. Andrews M, Edmunds M, Campbell A, Walls J, Feehally J. Systemic vasculitis in the 1980's - is there an increasing incidence of Wegener's granulomatosis and microscopic polyarteritis? J R Coll Physicians Lond 1990;24:284-288. [ Links ]
15. González-Gay MA, García-Porrúa C. Systemic vasculitis in adults in northwestern Spain 1988-97. Clinical and epidemiological aspects. Medicine (Baltimore) 1999;78:292-308. [ Links ]
16. Watts RA, González-Gay MA, Lane SE, Garcia-Porrúa C, Bentham G, Scott DG. Geoepidemiology of systemic vasculitis. Ann Rheum Dis 2001;60:170-172. [ Links ]
17. Lane SE, Watts RA, Bentham G, Innes NJ, Scott DG. Are environmental factors important in primary systemic vasculitis? A case-control study. Arthritis Rheum 2003;48:814-823. [ Links ]
18. Flint J, Morgan MD, Savage CO. Pathogenesis of ANCA-associated vasculitis. Rheum Dis Clin North Am 2010;36:463-477. [ Links ]
19. Xiao H, Heeringa P, Hu P, Liu Z, Zhao M, Aratani Y, et al. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Invest 2002;110:955-963. [ Links ]
20. Holle JU, Gross WL. ANCA-associated vasculitides: pathogenetic aspects and current evidence-based therapy. J Autoimmun. 2009;32:163-171. [ Links ]
21. Villiger PM, Guillevin L. Microscopic polyangiitis: Clinical presentation. Autoimmun Rev 2010;9:812-819. [ Links ]
22. Guillevin L, Durand-Gasselin B, Cevallos R, Gayraud M, Lhote F, Callard P, et al. Microscopic polyangiitis: clinical and laboratory findings in eighty-five patients. Arthritis Rheum 1999;42:421-430. [ Links ]
23. Savage CO, Winearls CG, Evans DJ, Rees AJ, Lockwood CM. Microscopic polyarteritis: presentation, pathology and prognosis. QJ Med 1985;56:467-483. [ Links ]
24. Hauer HA, Bajema IM, van Houwelingen HC, Ferrario F, Noël LH, Waldherr R, et al. Renal histology in ANCA-associated vasculitis: differences between diagnostic and serologic subgroups. Kidney Int 2002;61:80-89. [ Links ]
25. Levy JB, Hammad T, Coulthart A, Dougan T, Pusey CD. Clinical features and outcome of patients with both ANCA and anti-GBM antibodies. Kidney Int. 2004;66:1535-1540. [ Links ]
26. Collins CE, Quismorio FP Jr. Pulmonary involvement in microscopic polyangiitis. Curr Opin Pulm Med 2005;11:447-451. [ Links ]
27. Lauque D, Cadranel J, Lazor R, Pourrat J, Ronco P, Guillevin L, et al. Microscopic polyangiitis with alveolar hemorrhage. A study of 29 cases and review of the literature. Groupe d'Etudes et de Recherche sur les Maladies "Orphelines" Pulmonaires (GERM"O"P). Medicine (Baltimore) 2000;79:222-233. [ Links ]
28. Kawakami T, Soma Y, Saito C, Ogawa H, Nagahuchi Y, Okazaki T, et al. Cutaneous manifestations in patients with microscopic polyangiitis: two case reports and a minireview. Acta Derm Venereol 2006;86:144-147. [ Links ]
29. Kluger N, Pagnoux C, Guillevin L, Francès C; French Vasculitis Study Group. Comparison of cutaneous manifestations in systemic polyarteritis nodosa and microscopic polyangiitis. Br J Dermatol 2008;159:615-620. [ Links ]
30. Hattori N, Mori K, Misu K, Koike H, Ichimura M, Sobue G. Mortality and morbidity in peripheral neuropathy associated Churg-Strauss syndrome and microscopic polyangiitis. J Rheumatol 2002;29:1408-1414. [ Links ]
31. Furukawa Y, Matsumoto Y, Yamada M. Hypertrophic pachymeningitis as an initial and cardinal manifestation of microscopic polyangiitis. Neurology 2004;63:1722-1724. [ Links ]
32. Ku BD, Shin HY. Multiple bilateral non-hemorrhagic cerebral infarctions associated with microscopic polyangiitis. Clin Neurol Neurosurg 2009;111:904-906. [ Links ]
33. Pagnoux C, Mahr A, Cohen P, Guillevin L. Presentation and outcome of gastrointestinal involvement in systemic necrotizing vasculitides: analysis of 62 patients with polyarteritis nodosa, microscopic polyangiitis, Wegener granulomatosis, ChurgStrauss syndrome, or rheumatoid arthritis-associated vasculitis. Medicine 2005;84:115-128. [ Links ]
34. Nakamoto T, Yoshikawa M, Nakatani T, Yamane Y, Iwasawa S, Matsumoto M, et al. Microscopic polyangiitis that presented liver dysfunction prior to noted renal manifestations. Intern Med 2000;39:517-521. [ Links ]
35. Guillevin L, Pagnoux C, Teixeira L. Microscopic polyangiitis, in vasculitis. In: Ball G, Bridges S Jr, editors. Oxford (UK): Oxford University Press 2008;355-364. [ Links ]
36. Guillevin L, Pagnoux C, Seror R, Mahr A, Mouthon L, Le Toumelin P. French Vasculitis Study Group (FVSG). The Five-Factor Score revisited: assessment of prognoses of systemic necrotizing vasculitides based on the French Vasculitis Study Group (FVSG) cohort. Medicine 2011;90:19-27. [ Links ]
37. Pagnoux C, Mahr A, Hamidou MA, Boffa JJ, Ruivard M, Ducroix JP, et al. Azathioprine or methotrexate maintenance for ANCA-associated vasculitis. N Engl J Med 2008;359:2790-2803. [ Links ]
38. Guillevin L, Lhote F, Gayraud M, Cohen P, Jarrousse B, Lortholary O, et al. Prognostic factors in polyarteritis nodosa and Churg Strauss syndrome. A prospective study in 342 patients. Medicine 1996;75:17-28. [ Links ]
39. De Groot K, Harper L, Jayne DR, Flores Suarez LF, Gregorini G, Gross WL, et al. Pulse versus daily oral cyclophosphamide for induction of remission in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized trial. Ann Intern Med 2009;150:670-680. [ Links ]
40. Guillevin L, Cordier JF, Lhote F, Cohen P, Jarrousse B, Royer I, et al. A prospective, multicenter, randomized trial comparing steroids and pulse cyclophosphamide versus steroids and oral cyclophosphamide in the treatment of generalized Wegener's granulomatosis. Arthritis Rheum Dec 1997;40:2187-2198. [ Links ]
41. De Groot K, Rasmussen N, Bacon PA, Tervaert JW, Feighery C, Gregorini G, et al. Randomized trial of cyclophosphamide versus methotrexate for induction of remission in early systemic antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum 2005;52:2461-2469. [ Links ]
42. Hiemstra TF, Walsh M, Mahr A, Savage CO, de Groot K, Harper L, et al. European Vasculitis Study Group (EUVAS). Mycophenolate mofetil vs azathioprine for remission maintenance in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized controlled trial. JAMA 2010;304:2381-2388. [ Links ]
43. Jayne D, Rasmussen N, Andrassy K, Bacon P, Tervaert JW, Dadoniene J, et al. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. N Engl J Med 2003;349:36-44. [ Links ]
44. Mukhtyar C, Guillevin L, Cid MC, Dasgupta B, de Groot K, Gross W, et al. EULAR recommendations for the management of primary small vessel. vasculitis. Ann Rheum Dis 2009;68:310-317. [ Links ]
45. Jayne DR, Gaskin G, Rasmussen N, Abramowicz D, Ferrario F, Guillevin L, et al. European Vasculitis Study Group. Randomized trial of plasma exchange or highdosage methylprednisolone as adjunctive therapy for severe renal vasculitis. J Am Soc Nephrol 2007;18:2180-2188. [ Links ]
46. Richter C, Schnabel A, Csernok E, De Groot K, Reinhold-Keller E, Gross WL. Treatment of anti-neutrophil cytoplasmic antibody (ANCA)-associated systemic vasculitis with high-dose intravenous immunoglobulin. Clin Exp Immunol 1995;101:2-7. [ Links ]
47. Jayne DR, Chapel H, Adu D, Misbah S, O'Donoghue D, Scott D, et al. Intravenous immunoglobulin for ANCA-associated systemic vasculitis with persistent disease activity. QJM 2000;93:433-439. [ Links ]
48. Jayne DR, Davies MJ, Fox CJ, Black CM, Lockwood CM. Treatment of systemic vasculitis with pooled intravenous immunoglobulin. Lancet 1991;337:1137-1139. [ Links ]
49. Josselin L, Mahr A, Cohen P, Pagnoux C, Guaydier-Souquieres G, Hayem G, et al. Infliximab efficacy and safety against refractory systemic necrotising vasculitides: long-term follow-up of 15 patients. Ann Rheum Dis 2008;67:1343-1346. [ Links ]
50. Jones RB, Cohen Tervaert JW, Hauser T, Luqmani R, Morgan MD, Peh CA, et al. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med 2010;363:211-220. [ Links ]
51. Stone JH, Merkel PA, Spiera R, Seo P, Langford CA, Hoffman GS et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med 2010;363:221-232. [ Links ]

