










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Reumatología
Print version ISSN 0121-8123
Rev.Colomb.Reumatol. vol.18 no.2 Bogotá Apr./june 2011
PRESENTACIÓN DE CASO/CASE REPORT
2Reumatólogo. Hospital Universitario San Ignacio, Pontificia Universidad Javeriana, Bogotá, D.C., Colombia.
3Patóloga. Hospital Universitario San Ignacio, Pontificia Universidad Javeriana, Bogotá, D.C., Colombia.
Los autores declaran no presentar ningún conflicto de interés al momento de la redacción del manuscrito. No hubo ningún tipo de financiación.
Correspondencia: Lizet Paola Moreno Moreno, Oficina de Medicina Interna, Hospital Universitario San Ignacio, Carrera 7 N° 40-62, piso 7, Bogotá, D.C., Colombia. Teléfono: 5946161 Ext. 2340. Correo electrónico: lizet.moreno@javeriana.edu.co
Recibido: 19 de noviembre de 2010. Aceptado: 06 de mayo de 2011
Resumen
La enfermedad de Kikuchi Fujimoto se caracteriza histológicamente por la presencia de linfadenitis necrotizante, que igualmente se encuentra descrita en Lupus Eritematosos Sistémico, con características clínicas y patológicas comunes que pueden sugerir una posible relación entre estas dos enfermedades. ¿Es la enfermedad de Kikuchi Fujimoto una manifestación del Lupus Eritematoso Sistémico?. A continuación se presenta un caso de una mujer con Lupus Eritematoso Sistémico con linfadenitis necrotizante y linfadenopatía generalizada.
Palabras clave: linfadenitis necrotizante, lupus eritematoso sistémico, enfermedad de Kikuchi Fujimoto.
Summary
Kikuchi Fujimoto is histologically characterized by the presence of necrotizing lymphadenitis, which is also described in Lupus Erythematosus with common clinical and pathological features that may suggest a possible relationship between these two diseases. Is Kikuchi Fujimoto disease a manifestation of systemic lupus erythematosus? We report a case of a woman with Systemic Lupus Erythematosus with necrotizing lymphadenitis and lymphadenopathy.
Key words: necrotizing lymphadenitis, systemic lupus erythematosus, Kikuchi Fujimoto disease.
Introducción
La linfadenitis necrotizante puede estar asociada con múltiples condiciones, tales como neoplasias, infecciones y enfermedades inflamatorias1. Esta se ha descrito en linfomas tipo hodking y no hodking y en enfermedades infecciosas producidas por Yersenia Enterocolítica, Toxoplasma Gondii, Epstein Barr virus, linfogranuloma venéreo y VIH1.
La linfadenitis necrotizante histiocitica o enfermedad de Kikuchi Fujimoto (KFD) es extremadamente rara y su distribución es mundial. Se presenta con mayor prevalencia en Asia y principalmente en mujeres jóvenes2,3. Su etiología es desconocida y se ha encontrado una asociación entre KFD y Lupus Eritematoso Sistémico (LES), con características clínicas y patológicas comunes, que puede sugerir una posible relación entre estas dos enfermedades1,4-7. La enfermedad de Kikuchi Fujimoto es de carácter benigno y autolimitado, con resolución espontánea del cuadro entre 1 a 4 meses2.
A continuación se presenta un caso de una mujer con Lupus Eritematoso Sistémico con linfadenitis necrotizante y linfadenopatía generalizada.
Reporte de caso
Es una mujer de 26 años natural y procedente de Bogotá, Colombia, quien presenta cuadro de 2 meses de evolución de debilidad muscular de predominio proximal, asociado a artralgias simétricas en articulaciones de las manos con rigidez matinal de 30 minutos de duración, y pérdida de 7 kg de peso. Dos semanas antes del ingreso presenta exacerbación de los síntomas, asociado con fiebre de 38°C a 40°C, dolor abdominal difuso, emesis de contenido alimentario en tres ocasiones, epistaxis, hiporexia, astenia y adinamia. Estaba en tratamiento ambulatorio con prednisona 5 mg vía oral. Como antecedente de importancia tiene una tía con diagnóstico de artritis reumatoidea. Al examen físico de ingreso se encuentra con palidez mucocutánea generalizada con tensión arterial de 90/60, frecuencia cardiaca de 96, frecuencia respiratoria 20 con Sat O2 91%. Se evidencian múltiples adenopatías cervicales bilaterales, la mayor de 3 cm de diámetro y adenopatía axilar derecha del mismo tamaño. La auscultación cardiopulmonar era normal y presentaba dolor a la palpación en hipocondrio derecho sin signos de irritación peritoneal. Además debilidad proximal de predominio en cintura pélvica y escapular.
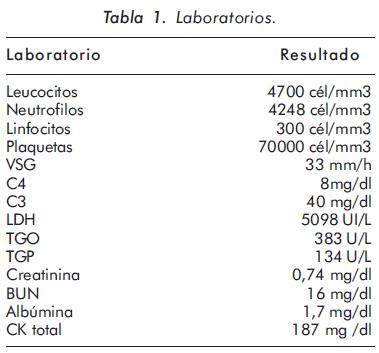
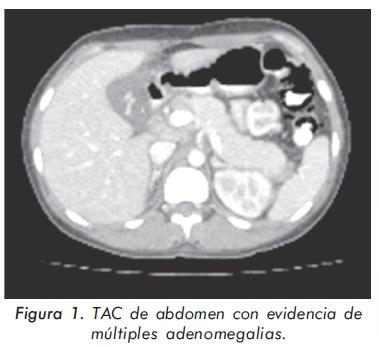
Los exámenes iniciales mostraron linfopenia, anemia normocítica, normocromica y trombocitopenia. La velocidad de sedimentación globular estaba aumentada, el complemento C4 y C3 consumido y LDH aumentada (tabla 1). Las pruebas de función hepática se encontraban alteradas con Transaminasas elevadas y las pruebas de función renal fueron normales. Se evidencia hipoproteinemia, con hipoalbuminemia y elevación de la CK total. La ecografía abdominal muestra adenomegalias retroperitoneales, intratoraccicas y periesplénicas. La Tomografía Axial Computarizada (TAC) de Abdomen (figura 1) evidenciaba un engrosamiento concéntrico vesicular sin otras alteraciones de la vía biliar, con múltiples adenomegalias paraaórtica e intraaortocavas. La TAC de tórax (figura 2) muestra un marcado engrosamiento de los septos inter e intralobulillares, en algunas áreas del pulmón zonas de opacidad en vidrio esmerilado y múltiples nódulos centrolobulillares.
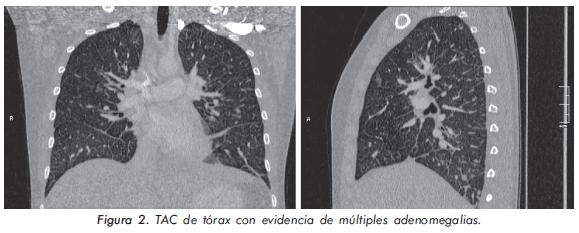
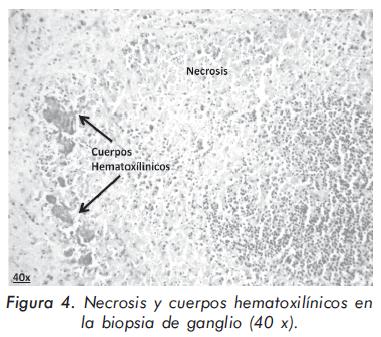
Se realizó biopsia de ganglio linfático cervical, la cual mostró una extensa necrosis del parénquima ganglionar, con mínimos folículos residuales; en la necrosis se identificaron detritos celulares, polvillo nuclear y abundantes cuerpos hematoxilínicos, que corresponden a fragmentos de DNA, con polisacáridos e inmunoglobulinas y que son característicos de la linfadenitis necrotizante asociada al Lupus Eritematoso Sistémico (figura 3 y 4).
Resultado de ANAS positivos 1/2560 con patrón homogéneo. Se realiza diagnóstico de LES y se inician pulsos de metil prednisolona, con mejoría de la sintomatología. Los síntomas mejoran rápidamente, sin presentación de nuevos picos febriles, mejoría objetiva de la fuerza de miembros superiores e inferiores y sin nuevos episodios de anemización. A pocos días de iniciar el tratamiento la paciente refiere disminución de la agudeza visual, con hallazgos al examen oftalmológico de exudados algodonosos en retina, compatibles con retinopatía lúpica.
Discusión
En el caso descrito, las manifestaciones principales fueron fiebre y una linfadenopatía sistémica, por lo que la sospecha inicial fue un linfoma. Posteriormente los estudios realizados, la anemia hemolítica, la trombocitopenia, la retinopatía lúpica, los ANAS elevados y la hipocomplementemia confirmaron el diagnóstico de Lupus Eritematoso Sistémico.
La linfadenopatía es un signo común en LES1,8. En varias series la prevalencia de linfadenopatía se ha encontrado en rangos de 12 a 59%1. El tamaño de los ganglios es de 0,5 hasta 3 a 4 cms y se presenta más frecuentemente a nivel cervical, mesentérico, axilar e inguinal8,9. Sin embargo la linfadenopatía generalizada es extremadamente rara en LES8,9, y más aún como primera manifestación de esta enfermedad1,8. Se ha descrito que cuando se presenta compromiso de tres o más nodos linfoides, las linfadenopatías son generalmente menores de un cm y solo se presentaron mayores a este tamaño en el 4,7% de los pacientes evaluados10.
La asociación de Lupus Eritematoso Sistémico con linfadenitis necrotizante histiocitica o enfermedad de Kikuchi Fujimoto (KFD) ha sido sugerida en varias revisiones1,4-7,9 y pueden ser patológicamente indistinguibles1. La etiología de ambas condiciones no es clara2. El diagnóstico de KFD se realiza a través de biopsia del nódulo linfoide comprometido, donde se encuentran áreas de necrosis cortical y paracortical, con presencia de cariorrexis, infiltrados histiocitarios y linfocitos con ausencia de neutrofilos4. Los estudios con microscopia electrónica han identificado estructuras reticulares tubulares en el citoplasma de linfocitos e histiocitos en pacientes con KFD4. Estas estructuras también han sido vistas en las células endoteliales y linfocitos de pacientes con lupus eritematoso sistémico y otros desordenes autoinmunes4.
Las características que pueden ser asociadas con LES y no con KFD son los cuerpos de hematoxilina, que se cree pueden representar núcleos degenerados que han reaccionado con anticuerpos antinucleares, y el fenómeno de azzopardi o incrustación de vasos sanguíneos con material nuclear4. Sin embargo en la KFD recientes estudios han demostrado células CD8 activadas después de infecciones virales, lo cual puede inducir apoptosis de linfocitos CD4 e hipotéticamente, los linfocitos apoptóticos podrían liberar antígenos nucleares y producir una respuesta autoinmune produciendo anticuerpos antinucleares11. Adicionalmente en la literatura se encuentra que el inmunofenotipo visto es virtualmente idéntico al de la KFD, incluyendo la presencia de patrón histiocítico, CD 68+ y mieloperoxidasa. Estos dos últimos corresponden a antígenos asociados con histiocitos4,12.
Todas las manifestaciones clínicas de la KFD pueden presentarse en el LES y algunos criterios diagnósticos del LES pueden estar presentes en la KFD13,14. En una revisión de casos de KFD se han descrito manifestaciones neurológicas, artritis, ulceras orales y se encontró la presencia de leucopenia en el 50% de los casos11.
Para explicar las características clínicas y patológicas ultraestructurales se ha planteado como hipótesis un evento común desencadenador, tales como un agente infeccioso o ambiental que puede producir ambos desórdenes1. Se sugiere la presencia de similitudes patogenéticas resultantes de la sobre reactividad inmunológica contra indeterminados factores etiológicos12. Sin embargo en la KFD los resultados de parámetros inmunológicos tales como factor reumatoideo y anticuerpos antinucleares, son en la mayoría de los casos negativos4, encontrándose en una revisión ANAS positivos solamente en el 7% de los casos reportados3. Alternativamente se ha planteado que KFD puede ser una linfadenitis necrotizante autoinmune que puede permanecer autolimitada o desarrollarse dentro de un LES y ser una manifestación inusual del mismo1,7.
Conclusión
Nosotros consideramos como hipótesis que la enfermedad de Kikuchi Fujimoto es una linfadenitis necrotizante histiocítica que puede presentarse dentro de un LES o ser una manifestación del mismo, y además que las diferencias presentadas entre estas dos enfermedades pueden ser secundarias a los procesos inmunológicos que pueden desencadenar una variante histológica del Kikuchi-Fujimoto, con presencia de cuerpos de hematoxilina, correspondiendo a una misma entidad con inmunofenotipo idéntico que puede dar sustento a esta teoría.
Agradecimientos
Al departamento de Medicina Interna y residentes del Hospital Universitario San Ignacio, especialmente al Dr. Erick Fabián Castaño por su participación en el estudio clínico.
Referencias
1. Eisner MD, Amory J, Mullaney B, Tierney L, Jr., Browner WS. Necrotizing lymphadenitis associated with systemic lupus erythematosus. Semin Arthritis Rheum. 1996;26(1):477-482. [ Links ]
2. Bosch X, Guilabert A. Kikuchi-Fujimoto disease. Orphanet J Rare Dis 2006;1:18. [ Links ]
3. Kucukardali Y, Solmazgul E, Kunter E, Oncul O, Yildirim S, Kaplan M. Kikuchi-Fujimoto disease: Analysis of 244 cases. Clin Rheumatol 2007; 26(1):50-54. [ Links ]
4. Bosch X, Guilabert A, Miquel R, Campo E. Enigmatic kikuchi-fujimoto disease: A comprehensive review. Am J Clin Pathol 2004;122(1):141-152. [ Links ]
5. Santana A, Lessa B, Galrao L, Lima I, Santiago M. Kikuchi-fujimoto's disease associated with systemic lupus erythematosus: Case report and review of the literature. Clin Rheumatol 2005;24(1):60-63. [ Links ]
6. Rao GS, Vohra D, Kuruvilla M. Is Kikuchi-Fujimoto disease a manifestation of systemic lupus erythematosus? Int J Dermatol 2006;45(4):454-456. [ Links ]
7. Mahajan T, Merriman RC, Stone MJ. Kikuchi-Fujimoto disease (histiocytic necrotizing lymphadenitis): Report of a case with other autoimmune manifestations. Proc (Bayl Univ Med Cent) 2007;20(2):149-151. [ Links ]
8. Kitsanou M, Andreopoulou E, Bai MK, Elisaf M, Drosos AA. Extensive lymphadenopathy as the first clinical manifestation in systemic lupus erythematosus. Lupus 2000;9(2):140-143. [ Links ]
9. Litwin MD, Kirkham B, Henderson DR, Milazzo SC. Histiocytic necrotising lymphadenitis in systemic lupus erythematosus. Ann Rheum Dis 1992;51(6):805-807. [ Links ]
10. Calguneri M, Ozturk MA, Ozbalkan Z, Akdogan A, Ureten K, Kiraz S, et al. Frequency of lymphadenopathy in rheumatoid arthritis and systemic lupus erythematosus. J Int Med Res 2003;31(4):345-349. [ Links ]
11. Sousa Ade A, Soares JM, de Santos MH, Martins MP, Salles JM. Kikuchi-fujimoto disease: Three case reports. Sao Paulo Med J 2010;128(4):232-235. [ Links ]
12. Hrycek A , Cieslik P, Szkrobka W, Pajak J. Kikuchifujimoto disease: A case report. Rheumatol Int 2005;26(2):179-181. [ Links ]
13. Hedia G, Jamel A, Maher A, Hanadi A, Agnes H, Nidhameddine K. Kikuchi-Fujimoto disease associated with systemic lupus erythematosus. J Clin Rheumatol 2005;11(6):341-342. [ Links ]
14. Gómez C, Eraso RM, Aguirre C, Perez M. Enfermedad de Kikuchi-Fujimoto: Presentación de un caso pediátrico. Biomédica 2010;30:465. [ Links ]

