










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Reumatología
Print version ISSN 0121-8123
Rev.Colomb.Reumatol. vol.18 no.3 Bogotá July/Sept. 2011
PRESENTACIÓN DE CASOS. REVISIÓN DE LA LITERATURA
Edgar Peñaranda-Parada1, Gerardo Quintana4, Federico Rondón H.5,
José Félix Restrepo6, Antonio Iglesias-Gamarra6
1Especialista en Medicina Interna, Residente de Reumatología segundo año, Universidad Nacional de Colombia.
2Especialista en Reumatología. Cali, Colombia.
3Especialista en Medicina Interna, Universidad Nacional de Colombia.
4Especialista en Reumatología. Profesor asistente Facultad de Medicina, Universidad Nacional de Colombia.
5Especialista en Reumatología. Profesor asociado Facultad de Medicina, Universidad Nacional de Colombia.
6Especialista en Reumatología, Profesor Titular Facultad de Medicina, Universidad Nacional de Colombia.
Los autores declaran no presentar ningún conflicto de interés al momento de la redacción del manuscrito.
Correspondencia: correo electrónico: nspinelb@gmail.com
Recibido: 17 de febrero de 2011 Aceptado: 8 de agosto de 2011
Resumen
Las enfermedades osteocondensantes son un grupo de patologías poco frecuentes que se caracterizan por aumento de la masa ósea, comprometiendo tanto a huesos largos como a huesos planos. Tradicionalmente, la radiología simple ha permitido su diagnóstico al identificar patrones de afectación ósea característicos de cada enfermedad. Actualmente, la caracterización molecular y genética ha facilitado la comprensión del sustrato fisiopatológico y la expresión fenotípica de estás patologías, sin embargo, la radiología simple continua teniendo un valor inconmesurable en el reconocimiento de las enfermedades osteocondensantes.
Palabras clave: osteoesclerosis, hiperostosis, enfermedades esclerosantes del hueso, huesos largos.
Summary
Sclerosing bone disorders are a rare group of diseases characterized by increased bone mass in both long and flat bones. Traditionally, plain radiography has allowed the diagnosis of these diseases identifying characteristic patterns of bone involvement. At present, the molecular and genetic characterization of these diseases has provided a better understand of their pathophysiology and phenotypic expression, however plain radiography continues to have an important role in the recognition of sclerosing bone disorders.
Key words: osteosclerosis, hyperostosis, sclerosing bone disorders, long bones.
Introducción
Las enfermedades osteocondensantes (EO) son un grupo de patologías raras que se caracterizan por un aumento en la masa ósea. El engrosamiento óseo afecta tanto al hueso trabecular (osteosclerosis) como al hueso cortical (hiperostosis), y puede ser generalizado o localizado. Estas enfermedades afectan los huesos originados a partir de los procesos de osificación endocondral y membranosa.
La osificación endocondral da origen a los huesos largos, el esqueleto axial, el esqueleto apendicular y la mandíbula, a partir de un molde de tejido cartilaginoso. Por su parte, la osificación membranosa ocurre a partir del tejido conjuntivo y forma los huesos planos. En la actualidad, las EO son reconocidas por sus manifestaciones clínicas, de laboratorio y en gran medida por sus hallazgos radiológicos. Sin embargo, la caracterización molecular y genética de estas patologías ha permitido comprender su fisiopatología, en donde las alteraciones en los procesos de resorción ósea y de neoformación de hueso, son los dos principales mecanismos relacionados con la expresión fenotípica de estas enfermedades.
Aquí presentamos dos casos de EO, describiendo sus características clínicas, de laboratorio y radiológicas, resaltando el compromiso de huesos largos. Además, se presenta una revisión de las más importantes EO de origen genético que afectan los huesos largos (osificación endocondral).
Caso 1
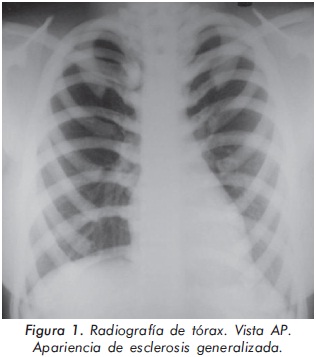
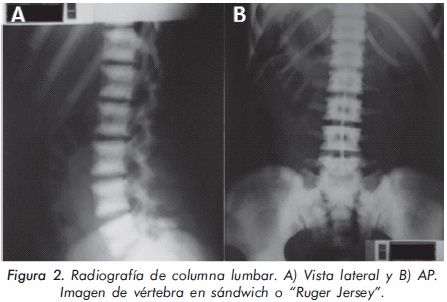
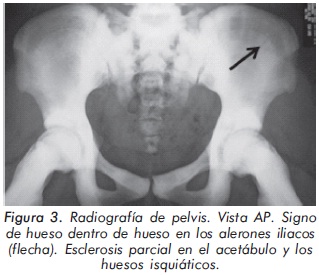
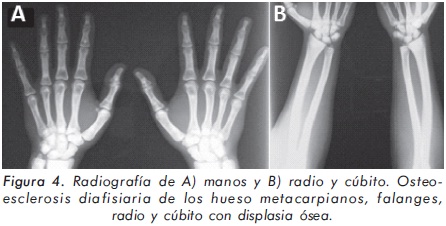
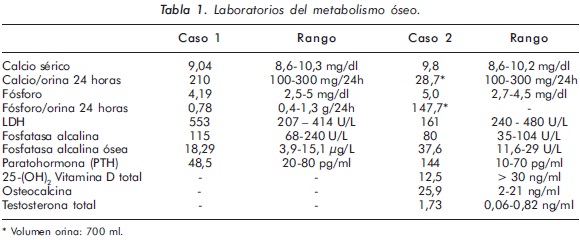
Mujer de 23 años de edad quien desde hace 7 años refiere dolores óseos en la columna cervical, cintura pélvica y miembros inferiores. El examen físico es normal a excepción de la presencia de torus palatino. El hemoleucograma y la química sanguínea son normales. La radiografía de tórax mostró aumento generalizado de la densidad ósea (Figura 1). En la radiografía de columna lumbosacra, las vértebras lumbares dan una apariencia de sándwich (Figura 2), con esclerosis densa y difusa adyacente a los platillos vertebrales superiores e inferiores, con falta de diferenciación corticomedular. La radiografía de pelvis mostró aumento de los alerones iliacos con un patrón geográfico de esclerosis densa con aspecto de "hueso dentro de hueso" (Figura 3). Las radiografías de huesos largos y manos mostraron osteoesclerosis y displasia ósea a nivel diafisiario y metáfisiario (Figuras 4 y 5). Además, se identificó esclerosis en la base del cráneo (Figura 6). La densidad mineral ósea (DMO) medida por densitometría ósea (DEXA) estaba aumentada, con un puntaje T de 9,9 (Z, 10,4) y 8,6 (Z, 8,9), en la columna lumbar y cuello femoral, respectivamente. El estudio metabólico del hueso mostró aumento leve de la fosfatasa alcalina específica del hueso (Tabla 1).
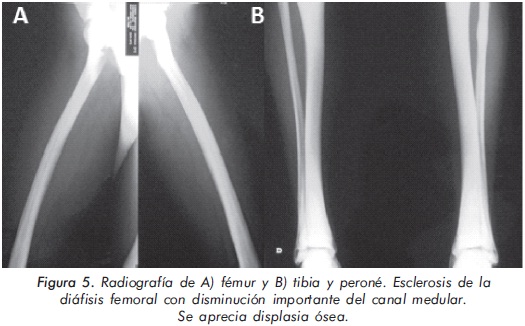
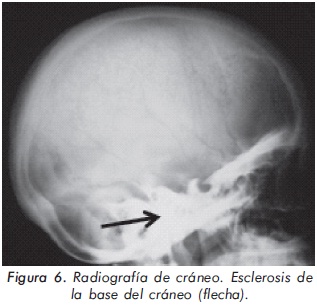
Actualmente, la paciente cursa con síntomas de radiculopatía lumbar secundarios a un canal lumbar estrecho.
Caso 2
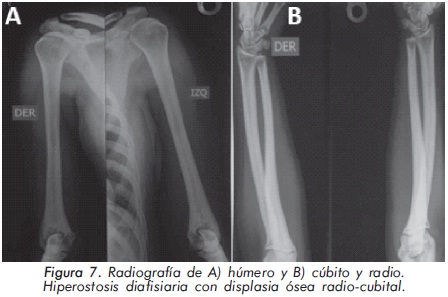
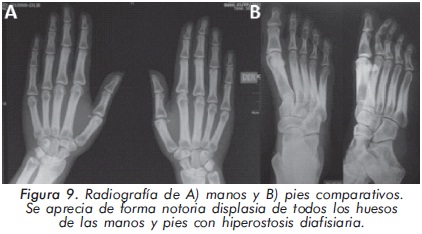
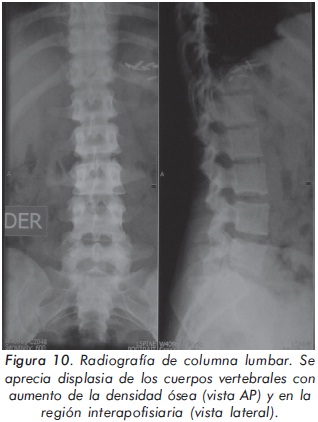
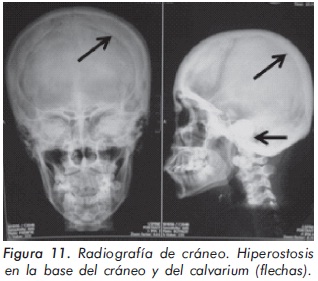
Mujer de 29 años de edad quien consulta al servicio de reumatología en julio de 2010 por dolor óseo generalizado, cervicalgia y artralgias de 4 meses de evolución. Los antecedentes médicosincluyen una insuficiencia renal crónica de etiología desconocida en hemodiálisis, hipertensión arterial y asma. Como único hallazgo al examen físico había dolor óseo generalizado a la digitopresión. Los anticuerpos antinucleares (ANAS) y los antígenos nucleares extractables (ENAS) fueron negativos. Como parte inicial del estudio del dolor óseo se realizó una serie de radiografías de huesos largos, la cual mostró esclerosis homogénea de las diáfisis, dado por engrosamiento de la cortical (hiperostosis) y disminución de la cavidad medular (Figuras 7 y 8).La apariencia ósea a nivel del radio, cúbito y fémur es displásica (Figuras 7 b y 8). Estos mismos hallazgos se encontraron en la radiografía comparativa de manos y pies (Figura 9). En la radiografía de columna lumbar, los cuerpos vertebrales son displásicos y hay esclerosis de las articulaciones interapofisiarias (Figura 10). A nivel del cráneo se encuentra esclerosis de la base y del calvarium (Figura 11). La DEXA reportó aumento de la DMO a nivel lumbar (1,617g/cm²) y del cuello femoral (1,116g/cm²), con un puntaje Z de 3,6 y 1,2, respectivamente. Los estudios del metabolismo óseo se presentan en la tabla 1.

Discusión
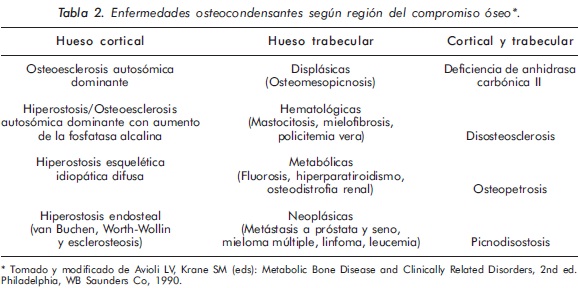
Las enfermedades osteocondensantes (EO) se caracterizan por aumento en la masa ósea, comprometiendo tanto a los huesos largos como a los planos1,2. En las EO la afectación de los huesos largos puede presentarse tanto en el hueso trabecular como en el hueso cortical. El engrosamiento óseo a nivel trabecular se denomina osteosclerosis, mientras que el engrosamiento que ocurre en la cortical del hueso se denomina hiperostosis. Además, los huesos largos pueden tener aspecto displásico o no3. Al considerar estos patrones de compromiso óseo en la radiología simple, junto con su localización anatómica y las manifestaciones clínicas y paraclínicas acompañantes, se puede orientar adecuadamente el tipo de patología que presenta el paciente (Tablas 2 y 3).
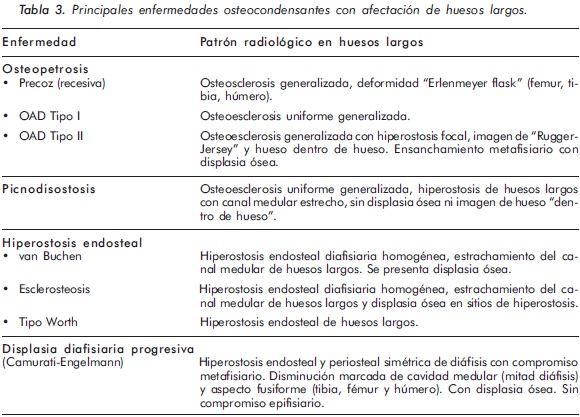
Teniendo en cuenta lo anterior, las características clínicas y radiológicas presentadas en el primer caso son consistentes con un diagnóstico de Osteopetrosis tipo II o enfermedad de Albert Schomberg. De las dos formas conocidas de osteopetrosis autosómica dominante (OAD) tipo I y II, la OAD tipo II es la forma más común con una prevalencia reportada de 5,5 casos por 100.000 personas4. A su vez, las formas autosómicas dominantes son más frecuentes que las recesivas (deficiencia de anhidrasa carbónica y mutación en el gen TCIRG1)5. En latinoamérica, y especialmente en Colombia, no se conoce la frecuencia de esta enfermedad, existiendo únicamente reportes de casos en la literatura6,7.
En la mayoría de casos de OAD los pacientes son asintomáticos, identificándose la enfermedad de forma incidental por medio de estudios radiológicos requeridos por otros motivos8. Radiológicamente, la ODA tipo II se manifiesta por osteoesclerosis segmentaria que compromete principalmente las placas terminales de las vértebras (vértebra en sandwich o imagen de ruger jersey), los alerones iliacos (imagen de "hueso dentro de hueso") y la base del cráneo. Estas formas de presentación radiológica son características de esta patología. El diagnóstico diferencial del compromiso axial se debe realizar principalmente con la osteomesopicnosis, la cual sólo compromete las vertebras lumbares9. Cuando el compromiso axial ocurre en todos los segmentos y ocasiona osteoesclerosis de todo el cuerpo vertebral se debe considerar la enfermedad de Van Buchem, Worth-Wollin, esclerosteosis y picnodisostosis9.
El 80% de los pacientes con OAD tipo II o enfermedad Albert Schomberg presenta fracturas no traumáticas de huesos largos, además de parálisis de pares creneanos, osteoartritis de cadera y osteomielitis de la mandíbula10. Sin embargo, en el 20% restante de estos pacientes las fracturas están ausentes al momento de la evaluación, como en el caso presentado. El compromiso de huesos largos en la ODA tipo II se caracteriza por osteoesclerosis generalizada con displasia ósea a nivel de las metáfisis y también en las diáfisis. En este caso, la DMO por DEXA se encontró aumentada, con puntajes T y Z elevados, consistente con las series de casos informadas en la literatura11,12.
El segundo caso corresponde a una enfermedad de van Buchem, y es un caso aislado dentro de la historia familiar. Esta patología también denominada hiperostosis cortical generalizada, es una displasia ósea autosómica recesiva que se caracteriza por un engrosamiento simétrico de la cortical de los huesos. La hiperostosis cortical compromete frecuentemente la mandíbula y el cráneo, afectando también las costillas y las diáfisis de los huesos largos, respetando las epífisis y metáfisis. Lo anterior concuerda con las imágenes presentadas de este caso, a excepción del compromiso mandibular que puede ser variable (Figuras 7 y 8). De igual forma se afectan los huesos tubulares de las manos y pies (Figura 9), y se puede apreciar compromiso de los cuerpos vertebrales y la región interapofisiaria por esclerosis, como se demuestra en la Figura 10. El estudio metabólico del hueso en este caso, da cuenta de normalidad en la fosfatasa alcalina, pero con ligero aumento de la fosfatasa alcalina ósea, un hallazgo consistente en la enfermedad de van Buchem, y previamente descrito también por Iglesias y cols en una serie de 4 pacientes con hiperostosis cortical generalizada13. El otro tipo de hiperostosis cortical generalizada es la variante Worth-Wollin, sin embargo difiere de la enfermedad de van Buchem, en el hecho que es autosómica recesiva, cursa con fosfatasa alcalina normal y tiene torus palatino13. Otra patología a considerar en el diagnóstico diferencial de la enfermedad de van Buchem es la informada por Hernandez-Cassis y cols, quienes describen la Hiperostosis/osteoesclerosis autosómica dominante con elevación de la fosfatasa alcalina. En está patología los hallazgos radiológicos son similares a la enfermedad de van Buchem, pero se diferencia en el patrón de herencia14. En la serie descrita por Iglesias y cols, los pacientes presentaron cefalea como síntoma inicial, sordera y en algunos hubo dolor osteomuscular en los miembros inferiores, este último síntoma compartido con el caso presentado. Además, la insuficiencia renal crónica presente en este segundo caso, que explica el estado de hiperparatiroidismo secundario a insuficiencia de vitamina D (Tabla 1), no se relaciona con el compromiso óseo de la paciente. Por lo tanto, no hay evidencia de enfermedad metabolica ósea secundaria a una osteodistrofía renal, la cual afecta predominantemente el hueso trabecular.
Las EO pueden ser adquiridas, iatrogénicas o tener un origen genético. Su diagnóstico no es facil, y se fundamenta en los estigmas clínicos y radiológicos de cada patología. Un importante grupo de estas enfermedades se presentan en la niñez y durante la pubertad, con deformidades óseas características que, junto con lás imágenes radiológicas, ayudan al diagnóstico. Por el contrario, en adultos con dolores óseos o fracturas patológicas en las extremidades o el esqueleto axial, se requeriere un alto índice de sospecha de estas entidades clínicas, indicándose la toma de imágenes de radiología simple. Lo anterior puede acercar al diagnóstico de estas patologías, siempre y cuando se hayan excluido otras enfermedades más prevalentes causantes de estos síntomas. La DEXA permite identificar, cuantificar y monitorizar el aumento de la DMO que ocurre en estas enfermedades. Además, algunos paraclínicos son de ayuda en la identificación de las principales causas adquiridas, metabólicas y iatrogénicas de estas enfermedades, como el hemoleucograma, creatinina sérica, velocidad de sedimentación globular, calcio, fósforo, fosfatasa alcalina específica del hueso, función hepática y electroforesis de proteínas15. En el diagnóstico diferencial de las EO se deben considerar las neoplasias sólidas con metástasis a hueso(ej: próstata y seno), las neoplasias hematológicas (ej: mieloma múltiple) y el hiperparatiroidismo secundario a insuficiencia renal crónica. La intoxicación por fluor y el tratamiento con bifosfonatos son las principales causas iatrogénicas, las cuales se caractarizan radiológicamente por osteoesclerosis15.
Respecto a las enfermedades osteocondensantes de origen genético, su estudio y entendimiento ha sido difícil debido a su rareza. La historia clínica y los hallazgos radiológicos han sido a lo largo de la historia los elementos más importantes para su diagnóstico. En 1997, el International Working Group on Constitutional Diseases of Bone subdividió las displasias esclerosantes del hueso en tres grupos: 1. Las enfermedades con aumento de la densidad ósea sin modificación de la forma del hueso; 2. Aquellas con incremento de la densidad ósea con compromiso diafisiario, y por último 3. Las enfermedades con incremento de la densidad ósea con compromiso metafisiario16.
Sin embargo, gracias al descubrimiento de las bases genéticas y moleculares de estas enfermedades, en la actualidad se pueden clasificar gran parte de las EO en base al mecanismo fisiopatológico subyacente. Los principales mecanismos fisiopatológicos son: 1. La disminución en la resorción ósea (actividad osteoclástica), 2. Aumento en la formación ósea (actividad osteoblástica), y 3. Mecanismos mixtos15.
Enfermedades osteocondensantes hereditarias con alteración en la actividad osteoclástica
Osteopetrosis
La osteopetrosis comprende varios subtipos de EO con un patrón de osteoesclerosis generalizada y engrosamiento del hueso cortical. Subtipos: a. Osteopetrosis maligna infantil autosómica recesiva; b. Osteopetrosis autosómica dominante o de inicio tardio (tipo I y II); c. Tipo intermedio y d) con acidosis tubular renal. Las formas de osteopetrosis autosómica dominante son las más prevalentes y por ende las más importantes. El principal mecanismo fisiopatológico reconocido en la osteopetrosis ocaciona una alteración en la resorción ósea osteoclástica, debido a un defecto en el proceso de acidificación en la laguna de resorción ósea. Por lo general, los parámetros bioquímicos de la homeostasis ósea no están alterados, sin embargo la fosfatasa ácida y la izoenzima de creatina (BB-CK) a menudo están elevados. En las formas malignas de la enfermedad (autosómica recesiva) puede existir hipocalcemia e hipoparatiroidismo secundario17.
Osteopetrosis maligna infantil autosómica recesiva: también llamada osteopetrosis de inicio precoz, se encuentra asociada a inactivación del gen TC1RG1, a mutaciones del gen ClCN7 (pérdida de la actividad del canal de cloro 7), defectos en el gen GL (OSTM) y a mutaciones inactivadoras del gen RANK-L, la cual se asocia a un número reducido de osteoclastos18,19. Este tipo de osteopetrosis se manifiesta al nacimiento y durante la infancia por ceguera, sordera y anemia severa. Esto es debido al crecimiento óseo excesivo que causa compresión de pares craneanos y disminución del espacio medular para la hematopoyesis. También presentan hepatoesplenomegalia (hematopoyesis extra-medular) e infecciones recurrentes. Radiológicamente hay aumento generalizado de la densidad ósea. Igualmente hay modelamiento óseo (displasia) en las terminales metafisiarias de los huesos largos, generando una deformindad denomidada "Erlenmeyer flask", la cual se caracterizada por un ensanchamiento de las metáfisis y diáfisis adyacentes con adelgazamiento de la cortical, principalmente en fémur, tibia y húmero.
Osteopetrosis autosómica dominante (OAD): es conocida como osteopetrosis "benigna" o de tipo adulto y tiene un patrón de herencia autosómica dominante. La mayoría de pacientes son asintomáticos, sin embargo ocasionalmente se reporta dolor óseo. Los hallazgos radiológicos se descubren incidentalmente permitiendo identificar dos fenotipos de esclerosis ósea8:
1. Osteopetrosis autosómica dominante tipo I: el gen relacionado con esta enfermedad esta localizado en el cromosoma 11q12-13. También se ha relacionado con mutaciones en el gen LRP5, el cual ocacionan aumento de la masa ósea, por lo tanto no es considerada propiamente como una enfermedad asociada a disminución de la resorción ósea. En esta condición las fracturas óseas son inusuales. Radiológicamente hay una osteoesclerosis uniforme generalizada principalmente en la bóveda craneana, columna vertebral y huesos largos20.
2. Osteopetrosis autosómica dominante tï-Schönberg: se relaciona con mutaciones heterocigotas desactivadoras del gen ClCN7. A diferencia de la OAD tipo I, la mayoría de pacientes presentan dolor óseo, osteomielitis y aumento del riesgo de fracturas. Las fracturas se presentan en el 80% de casos y ocurren principalmente en huesos largos (fémur) y arcos costales posteriores. Además hay escolisis y hasta en un 30% de los casos se presenta osteoartritis de cadera. Radiológicamente se caracteriza por un patrón de hiperostosis focal que a nivel de la columna vertebral da una imagen de vertebra en sandwich o "Rugger-Jersey". A nivel de los alerones iliacos se aprecia la imagen de "hueso dentro de hueso". También hay esclerosis de la base del cráneo21. El compromiso de huesos largos se caracteriza por un engrosamiento de la cortical en aproximadamente el 66% de los pacientes. De igual forma se puede apreciar aumento de las metáfisis femorales, dando la apariencia de deformidad "Erlenmeyer flask".
Picnodisostosis
Esta patología se origina por la deficiencia de la enzima lisosomal osteoclástica Catepsina K (cisteinil proteinasa). Esta enzima participa en la resorcion ósea degradando el colágeno en la interface osteoclasto-hueso22.
Clínicamente los pacientes presentan baja estatura, prominencia del cráneo, facies dismórfica y aumento del riesgo de fracturas. También presentan manos cuadradas y dedos cortos con uñas hipoplásicas, tórax estrecho con pectum excavatum, escolisis, aumento de la lordosis lumbar y genu valgo debido a fracturas recurrentes23. Radiológicamente hay osteosclerosis uniforme generalizada que se manifiesta en la ñinez y aumenta con la edad. También hay hiperostosis de huesos largos con canal medular estrecho, pero contrario a la osteopetrosis, no hay mayor modelado óseo ni imagen de hueso dentro de hueso. Otras características radiológicas son la esclerosis de la base del cráneo con márgenes orbitarios radiodensos. Hay cierre retardado de las suturas y fontanelas (anterior), ángulo mandibular obtuso, ausencia parcial del hioides e hipoplasia en los aspectos laterales de la clavícula. También se evidencia hipoplasia de huesos de la cara, senos paranasales y falanges distales por acrosteolisis o aplasia24,25.
Enfermedades osteocondensantes hereditarias con aumento en la formación ósea
A. Osteoesclerosis y hiperostosis con mutación en genes de la vía del factor de crecimiento transformante β (TGF-β)
Estas enfermedades se caracterizan por osteoesclerosis y hiperostosis debido a mutaciones en los genes de la vía del TGF-β. A este grupo pertenecen la Displasia Diafisiaria Progresiva (DDP) o enfermedad de Camurati-Engelmann y la Osteopoikilosis.
Displasia diafisiaria progresiva o enfermedad de Camurati-Engelmann: la DDP es una displasia osteocondensante con un patrón de herencia autosómico dominante. Es debida a mutaciones en el gen TGFB1, causantes de una activación prematura del TGF-β126. Esta alteración genética produce aposición progresiva de nuevo hueso en la superficie endosteal y periostial de los huesos largos. Afecta a los infantes manifestándose por dolor y debilidad muscular, fatiga y alteraciones en la marcha (cojera). Puede haber compromiso de los nervios craneales (pérdida de la audición). Los síntomas, la edad de inicio y el compromiso radiológico tienen un curso y progresión variable, y en algunos pacientes los síntomas remiten en la adultez. La DDP se caracteriza radiológicamente por un engrosamiento cortical (hiperostosis) simétrico y bilateral de las diáfisis de los huesos largos, con diseminación gradual a la metáfisis y algunos defectos del modelamiento óseo. Los huesos largos adoptan un aspecto fusiforme con disminución de la cavidad medular. Afecta principalmente la tibia y el fémur. El cráneo y la pelvis están comprometidos en un 54% y 63%, respectivamente. Con menor frecuencia se afectan el peroné, el húmero, el cúbito y el radio27. El principal diagnóstico diferencial de la DDP es la esclerosis diafisiaria múltiple hereditaria o enfermedad de Ribbing. Esta se caracteriza por un patrón de herencia autosómica recesiva. Inicia después de la pubertad con síntomas neuromusculares leves. Radiológicamente afecta solo los huesos largos de forma asimétrica y asincrónica28.
Osteopoikilosis: esta enfermedad se caracteriza por focos ovales o redondeados de osteoesclerosis que dan una apariencia radiológica de hueso moteado. Se ha relacionado con mutaciones desactivadoras del gen LEMD3, involucrado en la inhibición de las proteínas Smad, las cuales participan en la transducción de señales del receptor del TGF-β. La Osteo-poikilosis afecta principalmente las terminaciones de los huesos tubulares, la región metafisiaria de los huesos largos, y los huesos del tarso, carpo y pelvis. Esta asociada a la dermatofibrosis lenticularis diseminada, y en su diagnóstico diferencial están las metástasis a hueso de tumóres sólidos29.
B. Osteoesclerosis y hiperostosis con mutación en genes de la vía Wnt
Bajo la denominación de hiperostosis endosteales esta un grupo de 3 EO causadas por mutaciones en genes de la vía Wnt, que generan aumento en la actividad osteoblástica. Estas son la enfermedad de van Buchen, la esclerosteosis y la hiperostosis endosteal tipo Worth (Worth-Wollin). Estas enfermedades se caracterizan radiológicamente por hiperostosis endosteal (superficie medular) de forma simétrica y craneo-tubular (cráneo y huesos largos) con modelamiento óseo30.
Enfermedad de van Buchen y esclerosteosis
Estas dos EO tienen un patrón de herencia autosómico recesivo. La enfermedad de van Buchen se origina en una deleción de 52-Kb en el cromosoma 17q12 que afecta un aumentador corriente abajo del gen SOST31. Por su parte, la esclerosteosis se origina apartir de una mutación desactivadora del gen SOST32.
Como consecuencia de estas alteraciones genéticas, la vía Wnt favorece la diferenciación y proliferación del osteoblasto, gracias a la actividad de la proteína beta- catenina33.
La enfermedad de van Buchen (hiperostosis cortical generalizada) es el prototipo de hiperostosis endosteal. Además del compromiso del cráneo y huesos largos, su principal característica clínica es el aumento asimétrico progresivo de la mandíbula durante la pubertad. En la adultez la mandíbula se torna engrosada y con un ángulo aumentado. También se presenta parálisis de los nervios craneales, sordera y atrofia óptica, como consecuencia del estrechamiento de los agujeros craneales. El compromiso de huesos largos ocaciona dolor óseo a la presión y no hay aumento del riesgo de fractura.
La esclerosteosis o enfermedad de Truswell-Hansen es similar en sus aspectos clínicos y radiológicos a la enfermedad de van Buchen. Se diferencia de esta última por la presencia de sindactilia (entre el segundo y tercer dedo con displasia ungueal), talla alta y aumento del peso en los niños34.
Estas dos entidades se caracterizan radiológicamente por un engrosamiento cortical endosteal que genera una corteza diafisiaria densa y homogénea con estrechamiento del canal medular. Hay modelamiento de huesos largos y la actividad de la fosfatasa alcalina se encuentra aumentada. Además hay osteoesclerosis en la base del cráneo, los huesos faciales, vertebras, pelvis y costillas.
Hiperostosis endosteal (Tipo Worth)
Es un desorden autosómico dominante originado por una mutación activadora del gen LPR535,36. Esta mutación ocasiona un aumento del hueso trabecular y cortical, y a diferencia de la enfermedad de van Buchen y la esclerosteosis, el síndrome Worth tiene un curso benigno y no hay fracturas. Los pacientes afectados por esta condición presentan hiperostosis de huesos largos, aumento de la densidad del esqueleto axial y torus palatino. Durante la adolescencia se presenta aplanamiento de la frente y elongación mandibular con disminución del ángulo gonial.
Conclusión
En la actualidad, gracias al descubrimiento de las bases genéticas y moleculares de las EO, aunado a los hallazgos radiológicos característicos, el diagnóstico de estas patologías ha dejado de ser un desafío, a pesar de su rareza en la población general. Sin embargo, ante la falta de estudios genéticos en nuestro medio, el diagnóstico de las EO continúa basándose en el patrón radiológico de afectación ósea, en donde el compromiso de los huesos largos es una clave diagnóstica importante.
Referencias
1. de Vernejoul MC, Kornak U. Heritable sclerosing bone disorders: presentation and new molecular mechanisms. Ann N Y Acad Sci 2010;1192:269-277. [ Links ]
2. Mejía-Vallejo J, Calvo E, Restrepo J F, Iglesias-Gamarra A. Enfermedades osteocondensantes: una nueva visión clínico-radiológica soportada en la genética y la inmunoosteología. Revista Colombiana de Reumatología 2009;16(1):46-60. [ Links ]
3. Toro CE, Quintana MA, Restrepo J F, Quintana G, Rondón F, Sánchez Álvaro, Molina F, Iglesias-Gamarra A. Osteoesclerosis axiales. Propuesta para una nueva aproximación diagnóstica. Revista Colombiana de Reumatología 2007;14(1):44-52. [ Links ]
4. Bénichou OD, Laredo JD, de Vernejoul MC. Type II autosomal dominant osteopetrosis (Albers-Schönberg disease): clinical and radiological manifestations in 42 patients. Bone 2000;26:87-93. [ Links ]
5. Bollerslev, J. Autosomal dominant osteopetrosis: bone metabolism and epidemiological, clinical, and hormonal aspects. Endocr Rev 1989;10:45-67. [ Links ]
6. Granados Sandoval E, Martínez J, Zepeda R, Trejo A, Sandoval C, Barrón JC. Osteopetrosis (enfermedad de Albers-Schonberg): reporte de un caso y revisión clínica. Med Int Mex 2007;23(6):542-545. [ Links ]
7. Benavente S, Castillo A, Sánchez Y. Osteopetrosis: reporte de un caso en paciente de 20 años. Rev. Chil. Reumatol 2011;27(1):25-28. [ Links ]
8. Bollerslev J, Andersen PE Jr. Radiological, biochemical and hereditary evidence of two types of autosomal dominant osteopetrosis. Bone 1998;91:7-13. [ Links ]
9. Quintana G, Fernández A, Restrepo J F, Rojas A, Calvo E, Rondón F, Sánchez A, Forero E, Iglesias-G A. Osteomesopicnosis asociada a litiasis renal. Informe de un caso. Diagnóstico diferencial de las enfermedades osteoesclerosantes axiales. Revista Colombiana de Reumatología 2004;11(4):341-346. [ Links ]
10. Bollerslev, J. Osteopetrosis. A genetic and epidemiological study. Clin Genet 1987;31:86-90. [ Links ]
11. Waguespack SG, Hui SL, Dimeglio LA, Econs MJ. Autosomal dominant osteopetrosis: clinical severity and natural history of 94 subjects with a chloride channel 7 gene mutation. J Clin Endocrinol Metab 2007;92(3):771-778. [ Links ]
12. Kaste SC, Kasow KA, Horwitz EM. Quantitative bone mineral density assessment in malignant infantile osteopetrosis. Pediatr Blood Cancer 2007;48(2): 181-185. [ Links ]
13. Iglesias A, Hernández-Cassis C, Cueto J, Chichilla A, Velasco O, Mercado J, et al. Hiperostosis cortical generalizada. Presentación de cuatro casos. Descripción de una nueva variante. Salud Uninorte 1988;4-5:181-196. [ Links ]
14. Hernández-Cassis C, Vogel CK, Hernandez T P, Econs MJ, Iglesias M, Iglesias A, et al. Autosomal dominant hyperostosis/osteosclerosis with high serum alkaline phosphatase activity. J Clin Endocrinol Metab 2003;88(6):2650-2655. [ Links ]
15. de Vernejoul MC. Sclerosing bone disorders. Best Pract Res Clin Rheumatol 2008;22(1):71-83. [ Links ]
16. International Working Group on Constitutional Disease of Bone. International Nomenclature and Classification of the Osteochondrodysplasias. 1997. Am J Med Genet 1998;79:376-382. [ Links ]
17. Whyte M P, Chines A, Silva DP Jr, Landt Y, Ladenson JH. Creatine kinase brain isoenzyme (BB-CK) presence in serum distinguishes osteopetroses among the sclerosing bone disorders. J Bone Miner Res 1996;11:1438-1443. [ Links ]
18. Frattini A, Orchard PJ, Sobacchi C, Giliani S, Abinun M, Mattsson J P, et al. Defects in TCIRG1 subunit of the vacuolar proton pump are responsible for a subset of human autosomal recessive osteopetrosis. Nature Genetics 2000;25:343-346. [ Links ]
19. Frattini A, Pangrazio A, Susani L, Sobacchi C, Mirolo M, Abinun M, et al. Chloride channel ClCN7mutations are responsible for severe recessive, dominant, and intermediate osteopetrosis. The Journal of Bone and Mineral Research 2003;18:1740-1747. [ Links ]
20. Van Hul E, Mathysen D, Bollerslev J, Gram J, Van Hul W. Autosomal dominant osteopetrosis type I is genetically linked to the same region on human chromosome 11 as the high bone mass phenotype. J Bone Miner Res 2000;15:S260. [ Links ]
21. Van Hul W, Bollerslev J, Gram J, Van Hul E, Wuyts W, Benichou O, et al. Localization of a gene for autosomal dominant osteopetrosis Albers-Schonberg disease to chromosome 1p21. Am J Hum Genet 1997;61:363-369. [ Links ]
22. Gelb BD, Shi G P, Chapman HA, Desnick RJ. Pycnodysostosis, a lysosomal disease caused by cathepsin K deficiency. Science 1996;273:1236-1238. [ Links ]
23. Maroteaux P, Lamy M. La pycnodysostose. Presse Med 1962;70:999-1002. [ Links ]
24. Jacobson HG. Dense bone-too much bone: Radiological considerations and differential diagnosis (parts I and II). Skeletal Radiol 1985;13:1-20 y 97-113. [ Links ]
25. Edelson JG, Obad S, Geiger R, et al: Pycnodysostosis: Orthopedic aspects, with a description of 14 new cases. Clin Orthop 1992;280:263-276. [ Links ]
26. Janssens K, ten Dijke P, Ralston SH, Bergmann C, Van Hul W. Transforming growth factor-beta 1 mutations in Camurati-Engelmann disease lead to increased signaling by altering either activation or secretion of the mutant protein. J Biol Chem 2003;278(9):7718-7724. [ Links ]
27. Janssens K, Vanhoenacker F, Bonduelle M, Verbruggen L, Van Maldergem L, Ralston S, et al. Camurati-Engelmann disease: review of the clinical, radiological, and molecular data of 24 families and implications for diagnosis and treatment. Journal of Medical Genetics 2006;43:1-11. [ Links ]
28. Vanhoenacker FM, De Beuckeleer LH, Van Hul W, Balemans W, Tan GJ, Hill SC, De Schepper AM. Sclerosing bone dysplasias: genetic and radioclinical features. Eur Radiol 2000;10:1423-1433. [ Links ]
29. Hellemans J, Preobrazhenska O, Willaert A, Debeer P, Verdonk PC, Costa T, et al. Loss-of-function mutations in LEMD3 result in osteopoikilosis, Buschke-Ollendorff syndrome and melorheostosis. Nat. Genet 2004;36:1213-1218. [ Links ]
30. Krishnan V, Bryant HU, Macdougald OA. Regulation of bone mass by Wnt signaling. The Journal of Clinical Investigation. 2006;116:1202-1209. [ Links ]
31. Balemans W, Patel N, Ebeling M, Van Hul E, Wuyts W, Lacza C, et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet 2002;39(2):91-97. [ Links ]
32. Brunkow ME, Gardner JC, Van Ness J, Paeper BW, Kovacevich BR, Proll S, et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet 2001;68(3):577-589. [ Links ]
33. Krishnan V, Bryant HU, Macdougald OA. Regulation of bone mass by Wnt signaling. J Clin Invest 2006;116(5):1202-1209. [ Links ]
34. Hamersma H, Gardner J, Beighton P. The natural history of sclerosteosis. Clin Genet 2003;63(3):192-197. [ Links ]
35. Boyden LM, Mao J, Belsky J, Mitzner L, Farhi A, Mitnick MA, et al. High bone density due to a mutation in LDL-receptor-related protein 5. The New England Journal of Medicine 2002;346:1513-1521. [ Links ]
36. Little RD, Carulli J P, Del Mastro RG, Dupuis J, Osborne M, Folz C, et al. A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. American Journal of Human Genetics 2002;70:11-19. [ Links ]

