










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Colombiana de Reumatología
versión impresa ISSN 0121-8123
Rev.Colomb.Reumatol. v.18 n.4 Bogotá oct./dic. 2011
HISTORIA
1Fellow de Neurología, Hospital San Ignacio de Loyola. Chicago. EUA.
2Reumatología. Unidad de Reumatología, Universidad Nacional de Colombia.
3Profesor titular de Medicina Interna y Reumatología, Facultad de Medicina, Universidad Nacional de Colombia.
Correspondencia, Dr. Antonio Iglesias G.: iglesias.antonio1@gmail.com
Los autores declaran no presentar ningún conflicto de interés al momento de la redacción del manuscrito.
Recibido: 13 de junio de 2011 Aceptado: 11 de octubre de 2011
Resumen
En este escrito hacemos una completa revisión de la enfermedad de Devic, desde sus primeras descripciones por Eugene Devic, hasta el concepto actual, donde es considerada una neuromielitis óptica. Exponemos las diferentes formas de presentación de la neuromielitis óptica así como los hallazgos clínicos y de laboratorio que permiten diferenciar esta entidad de la esclerosis múltiple, tarea que muchas veces no es fácil de realizar por la semejanza en la presentación clínica. Sin embargo es importante su diferenciación porque el tratamiento y el pronóstico difieren entre estas dos enfermedades. No obstante, el descubrimiento de los anticuerpos IgG anti NMO, dirigidos contra los canales de agua de acuaporina 4 fueron fundamentales en tal diferenciación. Realizamos una mención especial sobre la enfermedad de Devic y el lupus y finalmente hacemos unas notas sobre el tratamiento disponible para esta patología.
Palabras clave: enfermedad de Devic, lupus eritematoso sistémico, neuromielitis óptica, esclerosis múltiple.
Summary
In this paper we do a complete review of Devic's disease, from its first descriptions by Eugene Devic, to the current concept, which is considered a neuromyelitis optica (NMO). We present the different forms of presentation of NMO as well as clinical and laboratory findings that distinguish this entity from multiple sclerosis, a task that frequently is not easy to perform because of the similarity in clinical presentation. Its differentiation is important because the treatment and prognosis differ between these two diseases. However the discovery of NMO IgG antibodies directed against aquaporin-4 water channels was a critical step in such differentiation. We made special mention of Devic's disease and lupus, and finally make some notes on the available treatment for this pathology.
Key words: Devic'disease, systemic lupus erythematosus, neuromyelitis optica, multiple sclerosis.
Introducción
La neuromielitis óptica, también conocida como enfermedad de Devic, es una enfermedad desmielinizante inflamatoria idiopática del sistema nervioso central que afecta preferencialmente los nervios ópticos y la medula espinal, típicamente respeta el cerebro y tiene un curso con recaídas.
En 1870, Sir Thomas Clifford Allbut1 expresó el interés de la ciencia al analizar la asociación de dos alteraciones neurológicas que se iniciaban en la médula espinal (mielitis), en cinco pacientes. Doce a trece semanas después los pacientes presentaron el compromiso ocular (neuritis). En 1889, Achard y Guinon2 describieron un estudio clínico-patológico de esta nueva entidad. Posteriormente Eugene Devic fue el primero en utilizar los términos de "neuromielitis óptica".

¿Quién fue Eugene Devic?
Después de describir la neuromielitis óptica, poco se sabía de este investigador de origen francés, hasta que Darío Giorgi3, médico del Departamento de Ciencias Oftalmológicas "La Sapienza" de la Universidad de Roma, averiguó la vida de este investigador.
Eugene Devic nació en Lyon el 24 de octubre de 18583; fue discípulo y colaborador del profesor Bouveret en Lyon. Se graduó de médico en 1886 con la tesis titulada "Des rachutes de la fièvre typhöde"3. En 1888, Devic empezó a trabajar como clínico en el Hospital de Antiquaille y posteriormente continuó su carrera en el servicio de Anatomía patológica entre 1892 y 1894 y luego trabajó en el Hospital de Lyon, donde le interesó la pediatría3.
Entre 1888 y 1891, publicó una serie de artículos en la revista médica de Lyon Province medicale sobre varias enfermedades de la infancia, como reumatismo nodoso, corea, varicela, tosferina, roséola, enuresis, tuberculosis del hígado y fiebre tifoidea. Con el doctor Bouveret publicó algunos artículos sobre gastroenterología (ver tabla 1, sobre las diferentes publicaciones desde 1892 a 1917)3.

Descripción de la neuromielitis óptica
De su producción científica, la neuromielitis óptica (NMO) fue su trabajo seminal; publicó dos artículos y con su estudiante Fernand Gault describieron diecisiete casos de neuromielitis óptica, que para esa época era un serie excelente4-6. Para el Congreso Francés de Medicina, en 1894 Devic escribía lo siguiente: "Ils représent une des nombreuses modalités cliniques constituées par les localisations multiples d'unmême temps divers points du système nerveux central et périphérique"5.
Esta hipótesis de Devic aún persiste. En 1897, Devic logró trabajar en cardiología y con el profesor Tripier escribió un capítulo sobre "Séméiologie da coeur et des vaisseaux" para el libro de patología del profesor Bouchard. Finalmente, un estudiante anónimo, el 23 de febrero de 1930, en la revista Lyon medicale, número 8, escribió lo siguiente:
Those who knew Dr. Devic only through his studies did not really know him. He was mostly himself while teaching the basic elements of the service he provided, first of all at the Red Cross Hospital and then at the Hospital of Lyon where he stayed until after World War I. There he educated an elite, an elite which has now dispersed, an elite which despite all still bears the imprint left by its master. He showed how to examine a patient, the patience needed and the method involved, the modesty, honesty and precision called for when making a diagnosis. He had a refined clinical sense and clarity of judgment which he made appears simple but that masked a broad education and culture. He was also highly conscientious, the greatest quality any worker can have and the worst vice of the ambitious. Eugènè Devic helped form a talented group of doctors, and had a major impact on medical training during his day. His greatest contribution was to have made clinical medicine into a school. It follows that for men like Eugènè Devic, who avoided all pomp, honors and awards that any judgment made of them mirrored their modest way of life. However, it is only right that they are remembered for standing out. Dr. Devic is dead. He requested a simple funeral. Throughout his life he avoided ostentation; I, more than anyone else, know he would have detested all this pomp and ceremony surrounding his name. Nonetheless, I do not feel I have betrayed his memory by recounting his life's work. He loved his science too much not to pardon one of his pupils, bound to him by numerous ties, for writing a brief summary in a medical journal o fall that he has left us3.
En 1914, Holden7 informa sobre cinco casos en los cuales la mielitis se complicó por una ceguera completa. En 1936, Balser8, después de estudiar a varios pacientes, pensaba que era parte de la esclerosis múltiple. Desde 1936, muchos observadores de diferentes continentes consideraban que la neuritis de Devic pudiese ser una forma de la esclerosis múltiple aguda, o de una encefalomielitis, y que era difícil establecer una diferenciación entres los tres grupos de entidades de acuerdo con los estudios de Russel Brain9 en 1929.
Desde 1936, el síndrome de Devic se asoció con una variante de la esclerosis múltiple, tratando de buscar una causa de la etiología de esta enfermedad.
Así, de esta manera, desde la descripción de Devic4-5 en 1894, y la tesis de Gault6 en 1894, la búsqueda de la etiología tuvo un gran interés en los diferentes investigadores como Goulden10 en 1914, Beck11 en 1927, Stansbury12 en 1949, y Mandler y cols.13 en 1993. En unos pocos casos la neuritis óptica y la mielitis se han asociado aisladamente a enfermedades como el lupus eritematoso sistémico14-16, neuro-Behcet17, miasteniagravis18, síndrome de Sjögren primario19, 20, tiroiditis de Hashimoto20 y varicela21, pero en la mayoría de los pacientes con enfermedad de Devic no se ha demostrado una asociación, y en muchos de estos casos se ha comprobado la presencia de anticuerpos antinucleares y anticuerpos dirigidos contra antígenos extraíbles, lo que sugiere una etiología autoinmune contra los órganos (nervio óptico y medula espinal). En algunas publicaciones interesantes como las de Beck11 en 1927, Stansbury12 en 1949, Ortiz de Zarate y cols.22 en 1968, Fukazawa y cols.23 en 1990, Mandler y cols.13 en 1993 y otras se planteaba que la enfermedad de Devic era una variante de la esclerosis múltiple. Uno de los trabajos seminales fue el de Leonardi y cols.24 en 1987, en el que realizaron un estudio neuropatológico donde observaron necrosis, cavitación y engrosamiento de la pared vascular a nivel del cordón espinal, además estudiaron el líquido céfaloraquídeo, y observaron que estos hallazgos no se observaban en la esclerosis múltiple; igual conclusión realizaron Mandler y cols.13. Otros estudios efectuados en el Reino Unido por Swingler y Compston en 1986 y 199025, 26 observaron que la enfermedad de Devic no era frecuente en el grupo étnico caucásico, como sí se observa en la esclerosis múltiple. A partir de los estudios de Miller y cols.27en 1987, Tashiro y cols.28 en 1987, Paty y cols.29 en 1988, Thompson y cols.30 en 1990 y Fasekas y cols. 31 en 1994 se demostró que la resonancia magnética del cordón espinal y el análisis del líquido céfaloraquídeo en los pacientes con enfermedad de Devic eran diferentes a lo observado en la esclerosis múltiple, y la banda oligoclonal que se advierte en el 90% de los pacientes con esclerosis múltiple se ha descrito solo en algunos pacientes con enfermedad de Devic.
Muchos pacientes con NMO pueden diagnosticarse como esclerosis múltiple, pero los estudios comparativos de Seze y cols.32, 33 en 2001 y 2003 plantean la posibilidad de que son dos enfermedades diferentes. Bergamaschi y Ghezzi34 un año después demuestran que la NMO puede asociarse con enfermedades autoinmunes y con la presencia de anticuerpos antinucleares, extractables, anticardiolipinas y anticélulas parietales.
Como muchas de las enfermedades autoinmunes no órgano-específicas y órgano-específicas, las infecciones y las alteraciones autoinmunitarias pueden estar comprometidas en la patogénesis de la NMO.
El nervio óptico y los cordones espinales pueden contener antígenos que son blancos por los procesos mediados por una respuesta inmunitaria, mediada por mecanismos humorales34-37. Al inicio del cuadro clínico, los eventos que comprometen los nervios ópticos y el cordón espinal suelen ocurrir en un mes o la crisis puede presentarse 120 meses después.
Ochenta a noventa por ciento de los pacientes con neuromielitis óptica tienen episodios de recaídas y se comprometen simultáneamente los nervios ópticos y la mielitis, más que un curso monofásico13,32,33, 37,38-43. Las recaídas suelen presentarse un año después en un 60% de los pacientes o al tercer año en el 90% de los pacientes.
Acuaporinas
En 1992, Preston y cols.44 identificaron el primer miembro de la familia de las acuaporinas. Actualmente se conocen once miembros en los mamíferos. Los canales de agua se han postulado como estructuras que permiten el paso del agua a través de ciertas membranas, y este es uno de los mecanismos para explicar la difusión plasmática41,44-46. Las acuaporinas se distribuyen ampliamente en el cuerpo, especialmente en los riñones, los eritrocitos, los pulmones y los epitelios secretorios como las glándulas salivares41,44-48. La acuaporina 4 (AQP4) se expresa en el sistema nervioso central, músculo esquelético, pulmones, riñones, estómago y glándulas exocrinas41,44-48.
La acuaporina-4 es la principal en los canales de agua del sistema nervioso central y se expresa en la membrana plasmática de los astrocitos; envuelven los capilares de sus alrededores, especialmente en los pies de los astrocitos. También se localiza en las membranas basolaterales de las células ependimarias ventriculares y en los núcleos hipotalámicos41,44-,47,48. La localización de la AQP4 permite la interacción con la distrofina, que incluye el alfa, sintrofina40,41,44-48. El papel importante de la AQP4 es la regulación de la distribución del agua a través del cerebro. La AQP4 participa en la manipulación del agua cerebral, por ello su papel en el edema cerebral secundario a tumores, trauma cerebral, abscesos cerebrales y accidentes cerebro-vasculares; esto se ha podido demostrar en ratones nulos para AQP4 (AQP4- null mice) sometidos a edema cerebral vasogénico48-51.
El nervio óptico y los cordones espinales tienen antígeno o autoantígeno de AQP4, que son blancos para los procesos inmunitarios mediados por vía humoral44, 47, 52-54. Estudios recientes demuestran que la AQP4 genera una respuesta celular T, que a la vez induce una activación de los linfocitos B específicos para AQP4, y una gran producción de anticuerpos anti-AQP4 y estos anticuerpos son los que se encuentran en los pacientes con neuromielitis óptica53,54-59 (ver espectro de la neuromielitis óptica). Experimentos recientes demuestran que los anticuerpos dirigidos contra la AQP4 son de la subclase IgG1 y tienen la capacidad de activar el complemento45,57,58. La transferencia pasiva de AQP4 intratecal de pacientes con NMO puede inducir lesiones como NMO en animales de experimentación45,58. Con los experimentos mencionados previamente, se explica el mecanismo humoral de la NMO, como una enfermedad órgano-específica y por ello se ha descrito en el lupus eritematoso sistémico, en el síndrome de Sjögren, en la miastenia gravis, en la tiroiditis de Haschimoto y además también se observa la presencia de otros anticuerpos como los ANAS, ENAS, anticar-diolipinas y anticuerpos anti-parietales44,47,52-59. Pero la AQP4 no se expresa en los oligoden-drocitos. Lennon y cols. 52,53 en 2004 y 2005 mostraron que los anticuerpos IgG anti- NMO se observan en pacientes con neuromielitis óptica y este anticuerpo podría ayudar a diferenciar a los pacientes con síndrome de Devic de las otras alteraciones desmielinizantes41-42,45,52-55,59-61.
Los anticuerpos anti-NMO-IgG se unen a la acuaporina-4 en los principales canales que regulan la homeostasis del agua en el sistema nervioso central44-47,52,53-63. Los anticuerpos IgG anti-NMO se pueden detectar en el suero de pacientes con algunos desórdenes relacionados a la neuromielitis, que incluye la esclerosis múltiple óptica-espinal de origen asiático; mielitis recurrente, asociada con mielitis longitudinal extensa (compromete más de tres cuerpos vertebrales); neuritis óptica aislada recurrente; neuritis óptica o mielitis en el contexto de ciertas enfermedades órgano-específicas (tiroiditis de Haschi-moto) y enfermedades no órgano-específicas como el lupus y el síndrome de Sjögren primario, lo que hoy en día se conoce como el espectro de patologías que producen la neuromielitis óptica44,52-63.
Un papel efector del anticuerpo IgG anti-NMO en la neuromielitis óptica no se ha podido documentar, aunado a una carencia de modelos en animales como sí ocurre con otras patologías neurológicas como la epilepsia y la enfermedad de Parkinson, lo cual pudiera explicar mejor los mecanismos informados (vida supra).
Criterios diagnósticos de la enfermedad de Devic
Las primeras propuestas elaboradas para el diagnóstico de la neuromielitis óptica fueron las de Mandler y cols.13 en 1993, en las que aplican los criterios clínicos y por primera vez se utilizan las neuroimágenes como la resonancia de la médula espinal y el análisis del LCR.
O'Riordan y cols.37 en 1996 utilizaron los mismos criterios para la NMO, pero incorporaron el concepto de monofásico o multifasico o recidivante.
Wingerchuk y cols.41 en 1999 analizaron los criterios absolutos, en los que incluyen la presencia de neuritis óptica, mielitis y la ausencia de enfermedad en otras áreas diferentes a los nervios ópticos y la mielitis. Introducen los criterios de soportes mayores y menores. En el 2006 estos criterios se revisan y se establece un consenso en la Clínica Mayo41-43,64-66.
Los criterios desarrollados por Dean M. Wingerchuk y cols. y luego revisados por los mismos expertos para la enfermedad de Devic son las guías para el diagnóstico de esta patología. Se requieren dos criterios absolutos y al menos otros dos o tres de soportes41-42,64-66.
- Criterios absolutos
- Neuritis óptica
- Mielitis aguda
- Criterios de soportes
Incluyen:
1. Resonancia magnética cerebral. Ausencia de esclerosis múltiple. En la NMO se caracteriza por la ausencia de lesiones parenquimatosas, pero pueden encontrarse algunas lesiones en la sustancia blanca que no sean características de la esclerosis múltiple.
2. Resonancia magnética del cordón espinal, especialmente las imágenes en T2 que comprometen más de tres segmentos de los cuerpos vertebrales, que corresponde a una lesión extensa, es decir una mielitis longitudinal27-35,37-42,64-73. La presencia de una lesión heterogénea en T2 sugiere necrosis o cavitación64-73.
3. Anticuerpos.
La seropositividad de los anticuerpos IgG anti-NMO. Estos anticuerpos evalúan la seropositividad de los anticuerpos anti-AQP4 por la técnica del radioinmunoensayo (ELISA)39,44,45,52,53,62.
Otros anticuerpos. Es necesario analizar otros anticuerpos como los ANAS, ENAS, anti-ADN de doble cadena, cardiolipinas, anti-TPO, anti-tiroglobulinas, especialmente cuando exista la posibilidad de una enfermedad auto-inmune como el LES o el síndrome de Sjögren primario. La verdadera incidencia no es conocida, pero pueden asociarse hasta en un 50% de los pacientes64-66.
4. Líquido céfalo raquídeo (LCR). Es importante el análisis del LCR como soporte para el diagnóstico. Ocasionalmente los pacientes tienen una pleocitosis con un incremento de las células blancas durante el episodio agudo o durante las exacerbaciones. En pacientes con neuro-mielitis aguda por lupus, el LCR se puede confundir con una meningitis bacteriana. El grado de celularidad es muy raro en la esclerosis múltiple. La banda oligoclonal generalmente es negativa en pacientes con enfermedad de Devic.
Ocasionalmente se pueden observar más de 50 glóbulos blancos/mm3 durante la exacerbación de la mielitis aguda, y en el recuento diferencial revela la presencia especialmente de neutrófilos y puede asociarse con una lesión severa de mielitis, como necrosis. Esta celularidad por neutrófilo es raro observarla en la esclerosis múltiple37,41-43,64-66,74,77,78.
En el 85% de los pacientes con esclerosis múltiple se pueden detectar bandas oligoclonales en el LCR; en cambio, se puede observar solo en el 15% a 35% de las series recientes de los pacientes con NMO. Incremento en la síntesis de IgG casi no se observa en la NMO41-43, 64-66.
5. Pruebas electro-diagnósticas. Los potenciales evocados visuales ocasionalmente pueden detectar lesiones subclínicas, tienen poco poder diagnóstico en la NMO.
El espectro de la neuromielitis óptica
Dean M. Wingerchuk y cols.42 en su publicación seminal de 2007, titulada The spectrum of neuromyelitis óptica, informan acerca de las diferentes variantes en las que se ha informado en los diferentes continentes las formas fenotípicas de la neuromielitis óptica.
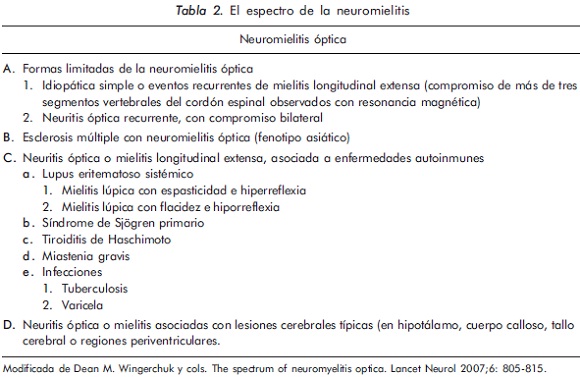
Neuromielitis óptica y el fenotipo de esclerosis múltiple de origen asiático
La esclerosis múltiple (EM) es una enfermedad inflamatoria desmielinizante del sistema nervioso central, que es desencadenada por una respuesta autoinmunitaria de tipo humoral, cuyo blanco importante es la mielina59.
La EM es rara en Asia, pero cuando aparece, compromete los nervios ópticos y el cordón espinal79. La EM óptico-espinal o EMOE tiene en su presentación algunas características clínicas parecidas a la enfermedad de Devic que se observa en la población occidental79. De acuerdo con los estudios de Lennon y cols.52, en el 2004, al demostrar como la inmunoglobulina IgG se une a algunos tejidos específicos del sistema nervioso central, pero específicamente en los pacientes con neuromielitis óptica (NMO) o NMO-IgG, se considera que pudiese ser una entidad diferente a la EM. Pero esta afirmación de Lennon y cols.52,53 no la pudieron corroborar Nakashima y cols.70 en el 2006, al estudiar pacientes japoneses con EM, pues informaron la presencia de NMO-IgG en el 60% de los pacientes con EMOE. Sin embargo, los anticuerpos contra NMO-IgG no se observan en todos los casos de NMO o de EMOE y se encuentran solamente en el 10% de los pacientes con EM clásica80. Esto pone de presente que establecer una dicotomía entre EM y NMO o enfermedad de Devic es complejo y existe una plausibilidad biológica, étnica y ambiental en la que puedan coexistir subgrupos de pacientes de EM y NMO que comparten algunas características clínicas, imagenológicas y de anticuerpos, que hagan difícil separar un cuadro clínico del otro, ya que cuando se describió la NMO, se informó que tenía un curso recidivante, convulsiones jacksonianas, cefalea, hipo severo, vómito y disartria, como lo informaron Devic3,4, Beck11, Balser y Stansbury12 y otros investigadores más recientes37,41-43,59,64-66,71,79,81.
Esclerosis múltiple, fenotipo asiático
Antes de la década de 1950, la EM se informaba esporádicamente en los países asiáticos. En 1958, Okinaka y cols.82 informaron que estudiaron 270 pacientes de EM diagnosticada entre 1890 y 1955, y observaron que el 65% de los pacientes era NMO, 24% tenían EM, 2% enfermedad de Schilder y otros casos no se pudieron clasificar. Entre los pacientes con NMO, 48% de los pacientes tenían un curso recidivante y los investigadores observaron por primera vez unos pacientes que tenían una forma intermedia entre EM y NMO82. A partir de este estudio, los japoneses denominaron NMO a la forma monofásica, es decir, aquellos casos en los que la enfermedad aparece simultáneamente comprometiendo los nervios ópticos y la mielitis, o cuando aparece la neuromielitis con un intervalo de menos de varias semanas, mientras que a los casos recidivantes los denominaron EM82.
En 1972, Kuroiwa y cols.83, en el Japón, en un estudio epidemiológico, revisaron 1.084 pacientes, y encontraron una baja prevalencia de EM. Esto le permitió a Shibasaki y cols.84 estudiar a la población japonesa y a pacientes de origen inglés y establecer una serie de características de la EM en la población asiática. Estas características clínicas son: 1) un compromiso severo de los nervios ópticos y del cordón espinal, 2) progresión rápida y 3) ocurrencia familiar rara, y no encontraron asociación a un HLA, pero plantearon la posibilidad de que la EM de origen asiático pudiese ser diferente a la que se observa en la población occidental. Kira y cols.59,79,85 tienen varias publicaciones sobre este tópico y plantean que existe una diferencia entre el EMOE de origen asiático y la EM convencional que se origina en occidente. Describen que en el Japón, entre el 15% y el 40% de los pacientes con EM tienen algunas características del fenotipo EMOE y se caracterizan por: 1) iniciación a una edad mayor, 2) más frecuencia en las mujeres, 3) muchas recaídas, 4) un compromiso severo en los nervios ópticos y en la médula espinal, con mucha discapacidad, 5) pocas lesiones cerebrales, 6) desarrollo de una mielitis longitudinal extensa en la resonancia magnética, 7) marcada pleocitosis con neutrofilia en el líquido cefalorraquídeo, 8) ausencia de bandas oligoclonales y 9) el HLA que se ha descrito es el HLA-DPB*0501, a diferencia de la variante occidental que tiene el HLA-DRBI*150159,85,86.
Histopatología
Desde las publicaciones de Beck17 en 1927 y de Stansbury12 en 1949 se empezó a describir los hallazgos anatomo-patológicos de la NMO y se notó la intensa desmielinización, la pérdida axonal y la proliferación de la microglia. Posteriormente los estudios de Okinaka y cols.82, Hung87, Tabira y cols.88 e Ikuta y cols.89, además de lo anotado más arriba, observaron infiltración linfocítica perivascular, proliferación vascular que se advierte en los nervios ópticos y en el cordón espinal generando una lesión necrosante y cavitaciones. De acuerdo con el estudio de Tabira y cols.88, quienes practicaron 90 autopsias de EM en el Japón desde 1955 a 1980, se observaron 25 pacientes con la forma clásica, 25 pacientes con enfermedad de Devic y 43 casos con la forma mixta; en esta última, notaron una severa desmielinización con necrosis tisular en los nervios ópticos y en el cordón espinal. Sin embargo, observaron algunas lesiones en el tronco cerebral y en el cerebro. Con estas evidencias histopatológicas es muy difícil diferenciar algunos casos de EM y NMO88.
Resonancia magnética de la médula espinal
La mielitis longitudinal que se observa a nivel de la médula espinal puede comprometer más de tres segmentos vertebrales, con poco compromiso cerebral; son las señales características que se observan en la NMO en los países occidentales, pero no se observan en la EM clásica, aunque sí en el fenotipo asiático, de acuerdo con los criterios de Barkhof90. Pittock y cols.91 informaron la presencia de lesiones cerebrales asintomáticas en la resonancia magnética cerebral en los pacientes con NMO. De acuerdo con estos mismos investigadores, las lesiones cerebrales se observan donde la acuaporina-4 (AQP4) es abundante, como en el diencéfalo adyacente al tercer ventrículo, en el puente y en el cerebelo adyacente al cuarto ventrículo79,91,92. En esta serie, el 10% de los pacientes con NMO-IgG positivos tenían compromiso cerebral, que era indistinguible de las lesiones de la EM, lo que genera una posible sobreposición entre la EM y la NMO.
Enfermedad de Devic y lupus
Deodhar y cols.93 informaron en 1999 el caso de un paciente que comprometió el cordón espinal de C3 a T2 y de T7 hasta el cono medular, que ellos describieron como el primer caso de mielitis longitudinal asociado a lupus. Posteriormente, Téllez-Zenteno y cols.94 describieron la primera serie de mielopatía asociada a lupus, que se clasificó como mielitis longitudinal. Describieron seis casos, en los que se observó una señal extensa en la resonancia magnética a nivel de T2, que comprometía varios segmentos del cordón espinal y torácico. Cuatro de los pacientes tenían anticuerpos anti-cardiolipinas, sugiriendo que podría tratarse de una característica de la mielitis longitudinal.
Birnbaum y Kerr95, del departamento de Neurología del Johns Hopkins, en el 2007, informaron el primer caso de anticuerpos IgG anti-NMO que confirmó el diagnóstico del síndrome de Devic en un paciente con lupus. Es interesante que el síndrome fuera diagnosticado durante los episodios recurrentes de mielitis longitudinal, antes de presentar el episodio de neuritis óptica. A este paciente con anticuerpos IgG anti-NMO con mielitis longitudinal se le administró una terapia inmunosupresora agresiva, para evitar los riesgos de las recaídas95.
Paira y cols.96 en el 2010 informan sobre otros dos pacientes con neuromielitis óptica con manifestaciones autoinmunitarias que sugieren lupus con títulos altos de anti IgG-NMO (títulos de 1/ 500). Los pacientes fueron tratados con cinco bolos de metilprednisolona de 1 g y 150 mg de azatioprina/día.
Pittock y cols.54, 55 evaluaron la presencia de autoanticuerpos en pacientes de Estados Unidos con NMO y mielitis transversa longitudinal extensa. Se detectaron anticuerpos antinucleares en el 43,8% y anticuerpos Ro en el 15,7%. Ambos tipos de anticuerpos se encontraron más frecuentemente en los pacientes con anticuerpos IgG anti-NMO positivos que en los pacientes con anti-IgG-NMO negativos54, 55.
Si no hay criterios para el diagnóstico de lupus o síndrome de Sjögren primario, la seropositividad para los anticuerpos antinucleares o para los anticuerpos Ro y La no excluye el diagnóstico para NMO39,54,55.
La frecuencia para la detección de anticuerpos IgG anti-NMO en pacientes pediátricos con NMO se encontró en el 76%39, 97.
Llama la atención que en el estudio de Pittock y cols.54 no se encontró un solo caso de anticuerpos IgG anti-NMO de los 49 controles con lupus y síndrome de Sjögren primario, pero ninguno de estos pacientes tenía compromiso de los nervios ópticos y mielitis54.
Informes aislados de pacientes con síndrome antifosfolipídico primario98,99-101, lupus95,99-107, síndrome de Sjögren primario109,110 y endocrinopatías autoinmunes111-113 muestran que actualmente cada vez son más frecuentes los casos con NMO con patología autoinmune, confirmando la naturaleza autoinmune y lo complejo e interesante de la NMO.
Mielitis y lupus
La mielopatía en el lupus es un síndrome inflamatorio grave que compromete el cordón espinal lo cual genera debilidad, cuadriparesia, adormecimiento y déficit a nivel de los esfínteres114. De acuerdo con Theodoridou y Settas115, la mielitis puede afectar entre el 1% y el 2% de los pacientes con LES. Este compromiso se considera mil veces más que la prevalencia de mielitis idiopática en la población general, según los informes de Kaplin y cols.116. La mayoría de los casos de mielitis en los pacientes con lupus son informes de casos. En los estudios de cohorte sobre mielitis en las enfermedades desmielinizantes se han descrito dos fenotipos diferentes como la esclerosis múltiple y la neuromielitis óptica (NMO)95,97. Birnbaum y Kerr, del Johns Hopkins, revisaron su cohorte de pacientes y analizaron 22 pacientes. Observaron que 11 pacientes tenían compromiso a nivel de la sustancia gris y estos pacientes consultaban por flacidez e hiporeflexia, mientras los otros 11 pacientes que tuvieron una disfunción de la sustancia blanca consultaron por espasticidad e hiporreflexia. Los pacientes con disfunción de la sustancia gris presentaron paraplejia irreversible con un curso monofásico o polifásico, asociada con una alta actividad del lupus. En los estudios del líquido cefalorraquídeo era difícil de diferenciarlo de una meningitis bacteriana. Previo a la paraplejia irreversible, estos pacientes consultaban por fiebre y retención urinaria, generalmente el cuadro clínico se confundía con una infección urinaria. El otro subtipo, en el que había disfunción de la sustancia blanca, tenía neuromielitis óptica y se asociaba más con la presencia de anticuerpos anticardiolipinas y anticoagulante lúpico95,106,107.
El anticuerpo IgG anti-NMO fue positivo en cuatro de siete pacientes con mielitis de la sustancia blanca. En cambio en los pacientes en los que se comprometió la sustancia gris, fue positivo en uno de ocho pacientes.
Ambos subtipos de mielitis recibieron una inmunosupresión a base de esteroides y ciclofos-famida. Pero a los pacientes en las que se observo una disfunción de la sustancia gris, además de recibir esteroides y ciclofosfamida, necesitaron plasmaferesis y gamaglobulinas95,106,107,114.
Tratamiento
Las recomendaciones terapéuticas son solo experiencias anecdóticas de pequeñas series de casos no controlados, debido al problema agudo que genera la neuromielitis óptica en la forma monofásica y en los eventos recidivantes. Los principales objetivos para el tratamiento de la NMO son controlar los ataques agudos, la prevención de las complicaciones médicas, la rehabilitación y tratar de prevenir los futuros ataques. Para los pacientes en la crisis monofásica y durante las recaídas agudas se utilizan 1000 mg de metilprednisolona por día, durante cinco días consecutivos39,41-43,64-66,95,106,107,114.
En aquellos pacientes que no respondan a los bolos de metilprednisolona, el uso de la plasmaferesis está indicado y se pueden utilizar siete recambios de 55 ml/Kg de peso117-123. El uso de las gamaglobulinas se ha practicado en algunos casos en forma anecdótica123. El compromiso medular puede generar una falla respiratoria que requiere que el tratamiento sea realizado en las unidades de terapia intensiva, para su soporte ventilatorio y vigilar el estado de una lesión bulbar. Además se requieren medidas médicas para prevenir el tromboembolismo pulmonar, la neumonía por aspiración, las infecciones urinarias y las úlceras por presión.
En la serie de Mandler y cols.13, una vez realizada la inducción de los casos agudos, en el mantenimiento del tratamiento, para evitar las recaídas, se utilizó un esquema basado en prednisona oral y azatioprina 2 mg/Kg/día13,120,124,125. La prednisona se va disminuyendo de acuerdo con el estado clínico, hasta una dosis de 10 mg/día. Los autores utilizaron la escala Expanded Disability Status para analizar la mejoría en los 18 meses de tratamiento39. También se han utilizado bolos de ciclo-fosfamida126 y mitoxantrone IV127. En pacientes con mielitis lúpica longitudinal y casos de síndrome de Sjögren primario y lupus con neuromielitis que no respondan al uso de metil prednisolona en bolos, plasmaferesis y gamaglobulinas, el uso del rituximab se ha hecho en forma anecdótica128-130.
Referencias
1. Allbut TC. On the ophthalmoscopic signs of spinal disease. Lancet 1870;1:76-78. [ Links ]
2. Achard and Guinon: Arch De mèd Expér 1889;1:696. [ Links ]
3. Giorgi D. Eugene Devic and the origin of neuromyelitis óptica. Eugène Devic el origine della neuromielite ottica. Riv Italiana di Neurobiología 2006;3-4:239-243. [ Links ]
4. Devic E. Myelíte aiguë dorse-lombaire avec névrite optique, autopsie. Congress Français Medicine (Premiere Session, Lyon) 1895;1:434-439. [ Links ]
5. Devic E. MyelÍte subaiguë compliquée de névrite optique. Bull Med 1894;8:1033-1034. [ Links ]
6. Gault F. De la neuromyélite optique aiguë. Thèse n°891, Faculté de Médecine et de Pharmacie de Lyon. [ Links ]
7. Holden: Arch. Ophth. 1914;43:231. [ Links ]
8. Balser: Brain, 1936;59:353. [ Links ]
9. Brain: Quart. J. Med. 1929-1930; 23:343. [ Links ]
10. Goulden C. Optic neuritis and mielitis. Optic neuritis and myelitis. Ophthalmic Review 1914;34:193-209. [ Links ]
11. Beck GM. A case of diffuse myelitis associated with optic neuritis. Brain 1927; 50: 687-703. [ Links ]
12. Stansbury FC. Neuromyelitis optica (Devic's disease). Presentation of five cases with pathological study and review of the literature. Arch ophtalmol 1949; 42: 292-335; 465-501. [ Links ]
13. Mandler RN, Davis LE, Jeffery DR, Kornfeld MK. Devis's neuromyelitis optica: a clinicophatological study of 8 patients. Ann Neurol 1993;34:162-168. [ Links ]
14. April RS, Vansonnenberg E. A case of neuromyelitis optica (Devic'syndrome) in systemic lupus erythe-matosus: clinicophatological report and review of the literature. Neurology 1976;26:1066-1070. [ Links ]
15. Kinney EL, Beroff RL, Rao NS, Lay M F. Devic's syndrome and systemic lupus Erythematosus. A case report and review of the literature. And Arch Neurol 1979;36:643-644. [ Links ]
16. Tola MR, Granieri E, Caniatti L, Paolino E, Monetti C, Dovigo L, et al. Systemic lupus Erythematosus presenting with neurological disorders. J Neurol 1992;239:61-64. [ Links ]
17. Motumara S, Tabira T, Kuroiwa Y. A clinical comparative study of multiple sclerosis and neuro-Behcet's syndrome. Journal of Neurology, Neurosurgery and Psychiatry 1980;43(3):210-213. [ Links ]
18. Goldman M, Herode A, Borenstein S, Zanen A. Optic neuritis, transverse myelitis and anti-DNA antibodies nine years after thymectomy for myasthenia gravis. Arth Rheum 1984;27:701-703. [ Links ]
19. Kahlenberg JM. Neuromyelitis Optica Spectrum Disorder as an Initial Presentation of Primary Sjögren's Syndrome. Neurology 2011:343-348. [ Links ]
20. Kahlenberg JM. Neuromyelitis Optica Spectrum Disorder as an Initial Presentation of Primary Sjögren's Syndrome. Neurology 2011:343-348. [ Links ]
21. Al-Deeb SM, Yaqub BA, Koja WO. Devic's neuromyelitis optica and varicella. J Neurol 1993;240:450-451. [ Links ]
22. Ortiz de Zarate JC. Tamaroff L, Sica REP, Rodriguez JA. Neuromyelitis óptica versus subacute necrotic myelitis: part II. Anatomical study of two cases. F Neurosurg Psychiatry 1968;31:641-645. [ Links ]
23. Fukazawa T, Hamada T, Tashiro K, Moriwaka F, Yanagihara T. Acute transverse myelitis in multiple sclerosis. J Neurol Sci 1990;100:217-222. [ Links ]
24. Leonardi A, Arata L, Farinelli M, Cocito L, Schenone A, Tabaton M, et al. Cerebrospinal fluid and neurophatological study in Devic's syndrome. Evidence of intrathecal immune activation. J Neurol Sci 1987;82:281-290. [ Links ]
25. Swingler RJ, Compston DAS. The distribution of multiple sclerosis in the United Kingdom. J Neurosurg Psychiatry 1986;49:1115-1124. [ Links ]
26. Compston DAS. Risk factors for multiple sclerosis: race or place? J Neurosurg Psychiatry 1990;53:821-823. [ Links ]
27. Miller DH, McDonald WI, Blumhardt LD, du Boulay GH, Halliday AM, Johnson G, et al. Magnetic resonance imaging in isolated non-compressive spinal cord syndromes. Ann Neurol 1987;22:714-723. [ Links ]
28. Tashiro K, Ito K, Maruo Y, Homma S, Yamada T, Fujiki N, et al. MR imaging of spinal cord in Devic disease. J Comput Assist Tomogr 1987;11:516-517. [ Links ]
29. Paty DW, Oger JJ, Kastrukoff L F, Hashimoto SA, Hooge J P, Eisen AA, et al. MRI in the diagnosis of multiple sclerosis: prospective comparison of clinical evaluation, EPs, Oligoclonal banding and CT. Neurology 1988;38:180-185. [ Links ]
30. Thompson AJ, Kermode AG, MacManus DG, Kendall BE, Kingsley D P, Moseley I F, et al. Patterns of disease activity in multiple sclerosis: clinical and magnetic resonance imaging study. BMJ 1990;300:631-634. [ Links ]
31. Fazekas F, Offenbacher H, Strasser-Fuch S. MRI of neuromyelitis optica: evidence for a distinc entity. J Neurol Neurosurg Psychiatry 1994;59:1140-1142. [ Links ]
32. de Seze J, Stojkovic T, Breteau G, Lucas C, Michon-Pasturel U, Gauvrit JY, et al. Acute myelopathies. Clinical, laboratory and outcome profiles in 79 cases. Brain 2001;124:1509-1521. [ Links ]
33. de Seze J, Lebrun C, Stojkovic T, Ferriby D, Chatel M, Vermersch P. , et al. Is Devic's neuromyelitis optica a separate disease? A comparative study with multiple sclerosis. Mult Scler 2003;9:521-525. [ Links ]
34. Bergamaschi R, Ghezzi A. Devic's neuromyelitis optica: clinical features and prognostic factors. Neurol Sci 2004;25:s364-s367. [ Links ]
35. Lucchinetti C F, Mandler RN, McGavern D, Bruck W, Gleich G, Ransohoff RM, et al. A role for humoral mechanisms in the pathogenesis of Devic's neuro-myelitis optica. Brain 2002;125:1450-1461. [ Links ]
36. Weinshenker BG, Wingerchuk DM, Nakashima I, Fujihara K, Lennon VA. OSMS in NMO, but not MS: proven clinically and pathologically. Lancet Neurol 2006;5:110-111. [ Links ]
37. O'Riordan JI, Gallagher HL, Thompson AJ, Howard RS, Kingsley D P, Thompson EJ, et al. Clinical, CSF, and MRI findings in Devic's neuromyelitis optica. J Neurol Neurosurg Psychiatry 1996;60:382-387. [ Links ]
38. Déral-Stéphant V, Roux-Lelièvre C, Vignal R, Stéphant E, Faivre A, Alla P . Neuromyélite optique de Devic : discussion diagnostique après dix ans d'évolution d'une neuropathie optique bilatérale sévère. J Fr Ophtalmol 2008;31:705-709. [ Links ]
39. Sellner J, Boggild M, Clanet M, Hintzen RQ, Illes Z, Montalban X, Du Pasquier RA, Polman CH, Sorensen PS, Hemmer B. EFNS guidelines on diagnosis and management of neuromyelitis optica. European J Neurol 2010;17:1019-1032. [ Links ]
40. Nakajima H, Hosokawa T, Sugino M, Kimura F, Sugasawa J, Hanafusa T, Takahashi T. Visual field defects of optic neuritis in neuromyelitis optica compared with multiple sclerosis. BMC Neurol 2010 Jun 18;10:45. [ Links ]
41. Wingerchuk DM, Hogancamp W F, O'Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic's Syndrome). Neurology 1999;53:1107-1114. [ Links ]
42. Wingerchuk DM, Lennon V, Lucchinetti C F, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Neurology 2007;6:805-815. [ Links ]
43. Wingerchuck DM, Weinshenker BG. Neuromyelitis optica: clinical predictors of a relapsing course and survival. Neurology 2003;60:848-853. [ Links ]
44. Preston GM, Carroll T P, Guggino WB, Agre P. Appearance of water channels in Xenopus oocytes expressing red cell CHIP28 protein. Science 1992;256:385-387. [ Links ]
45. Marignier R, Giraudon P, Vukusic S, Confavreux C, Honnorat J. Anti-aquaporin-4 antibodies in Devic's neuromyelitis optica: therapeutic implications. The Adv Neurol Disord 2010;3:311-321. [ Links ]
46. Kalluri SR, Rothhammer V, Staszewski O, Srivastava R, Petermann F, Prinz M, Hemmer B, Korn T. Functional Characterization of Aquaporin-4 Specific T Cells: Towards a Model for Neuromyelitis Optica. Plos One 2001;6:1-11. [ Links ]
47. Verkman AS Aquaporins: translating bench research to human disease. Journal of Experimental Biology 2009;212:1707-1715. [ Links ]
48. Amiry-Moghaddam M, Ottersen O P. The molecular basis of water transport in the brain. Nat Rev Neurosci 2003;4:991-1001. [ Links ]
49. Papadopoulos MC, Verkman AS, Papadopoulos MC, Verkman AS: Aquaporin-4 and brain edema. Ped Nephrol 2007;22:778-784. [ Links ]
50. Aoki K, Uchihara T, Tsuchiya K, Nakamura A, Ikeda K, Wakayama Y. Enhanced expression of aquaporin 4 in human brain with infarction. Acta Neuropathol 2003;106:121-124. [ Links ]
51. Papadopoulos MC, Saadoun S, Binder DK, Manley GT, Krishna S, Verkman AS: Molecular mechanisms of brain tumor edema. Neuroscience 2004;129:1011-1020. [ Links ]
52. Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti C F, Fujihara K, et al. A serum auto-antibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 2004;364:2106-2112. [ Links ]
53. Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin 4 water channel. J E sp Med 2005; 202:437-477. [ Links ]
54. Pittock SJ, Lennon VA, de Seze J, Vermersch P, Homburger HA, Wingerchuck DM, et al. Neuromyelitis optica and non organ-specific autoimmunity. Arch Neurol 2006;63:964-968. [ Links ]
55. Pittock SJ, Weinshenker BG, Lucchinetti C F, Wingerchuck DM, Corboy JR, Lennon VA. Neuromyelitis optica brain lesions localized at sites of high aquaporin 4 expression. Arch Neurol 2006;63:964-968. [ Links ]
56. Roemer S F, Parisi JE, Lennon VA, Benarroch EE, Lassmann H, Bruck W, et al. Pattern specific loss of aquaporin-4 immunoreactivity distinguishes neuromyelitis optica from multiple sclerosis. Brain 2007;130:1194-1205. [ Links ]
57. Bradl M, Misu T, Takahashi T, Watanabe M, Mader S, Reindl M, et al. Neuromyelitis optica: Pathogenicity of patient immunoglobulin in vivo. Ann Neurol 2009;66:630-643. [ Links ]
58. Saadoun S, Waters P, Bell BA, Vicent A, Verkman AS, Papadopoulos MC. Intracerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain 2010;133:349-361. [ Links ]
59. Kira J. Neuromyelitis Optica and Asian Phenotype of Multiple Sclerosis. Ann N Y Acad Sci 2008;1142:58-71. [ Links ]
60. Perwaiz MJ, Hammoudeh F, Schmidt F. Devic Syndrome: A Diagnostic Dilemma. J Natl Med Assoc 2011;103:176-178. [ Links ]
61. Misu T, Fujihara K, Kakita A, Konno H, Nakamura M, Watabane S, et al. Loss of aquaporin 4 in lesions of neuromyelitis optica: distinction from multiple sclerosis. Brain 2007;130:1224-1234. [ Links ]
62. Takahashi T, Fujihara K, Nakashima I, Misu T, Miyazawa I, Nakamura, et al. Anto-aguaporin-4 antibody is involved in the pathogenesis of NMO: a study on antibody titre. Brain 2007;130:1235-1243. [ Links ]
63. Lalive PH, Mengue T, Barman I, Cree BA, Genain C P. Identification of new serum autoantibodies in neuromyelitis optica using protein microarrays. Neurology 2006;67:176-177. [ Links ]
64. Wingerchuck DM, Lennon VA, Pittock SJ, Lucchinetti C F, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology 2006;66:1485-1489. [ Links ]
65. Mayo clinic. Devic's Disease Symptoms. www.mayoclinic.org/devic-disease/symptoms.html. Accessed april 14, 2010. [ Links ]
66. Matiello M, Jacob A, Wingerchuk DM, Weinshenker BG. Neuromyelitis optica. Curr Opin Neurol 2007;20:255-260. [ Links ]
67. Ghezzi A, Bergamaschi R, Martinelli V, Trojano M, Tola MR, Merelli E, et al. Clinical characteristics, course and prognosis of relapsing Devic's neuro-myelitis optica. J Neurol 2004;251:47-52. [ Links ]
68. Krampla W, Aboul-Enein F, Jecel J, Lang W, Fertl E, Hruby W, et al. Spinal cord lesions in patients with neuromyelitis optica: a retrospective long-term MRI follow-up study. Eur Radiol 2009;19:2535-2543. [ Links ]
69. Filippi M, Rocca MA, Moiola L, Martinelli V, Ghezzi A, Capra R, et al. MRI and magnetization transfer imaging changes in the brain and cervical cord of patients with Devic's neuromyelitis optica. Neurology 1999;53:1705-1710. [ Links ]
70. Nakashima I, Fujihara K, Miyazawa I, Misu T, Narikawa K, Nakamura M, et al. Clinical and MRI features of Japanese patients with multiple sclerosis positive for NMO-IgG. J Neurol Neurosurg Psychiatry 2006;77:1073-1075. [ Links ]
71. Sellner J, Lüthi N, Schüpbach WM, Gebhardt A, Findling O, Schroth G, et al. Diagnostic workup of patients with acute transverse myelitis: spectrum of clinical presentation, neuroimaging and laboratory findings. Spinal Cord 2009;47:312-317. [ Links ]
72. Cabrera-Gómez J, Saiz-Hinarejos A, Graus F, González-Quevedo A, Rodríguez-Rojas R, Quevedo-Sotolongo L, et al. Brain magnetic resonance imaging findings in acute relapses of neuromyelitis optica spectrum disorders. Mult Scler 2008;14:248-251. [ Links ]
73. Li Y, Xie P, Lv F, Mu J, Li Q, Yang Q, et al. Brain magnetic resonance imaging abnormalities in neuromyelitis optica. Acta Neurol Scand 2008; 118:218-225. [ Links ]
74. de Seze J, Stojkovic T, Ferriby D, Gauvrit JY, Montagne C, et al. Devic's neuromyelitis optica: clinical, laboratory, MRI and outcome profile. J Nuerol Sci 2002;197:57-61. [ Links ]
75. Bichuetti DB, Rivero RL, Oliveira DM, Souza NA, Abdala N, Oliveira EM, et al. Neuromyelitis optica: brain abnormalities in a Brazilian cohort. Arq Neuropsiquiatr 2008;66:1-4. [ Links ]
76. Milano E, Di Sapio A, Malucchi S, Capobianco M, Bottero R, Sala A, et al. Neuromyelitis optica: importance of cerebrospinal fluid examination during relapse. Neurol Sci 2003;24:130-133. [ Links ]
77. Bergamaschi R, Tonietti S, Franciotta D, Candeloro E, Tavazzi E, Piccolo G, et al. Oligoclonal bands in Devic's neuromyelitis optica and multiple sclerosis: differences in repeated cerebrospinal fluid examinations. Mult Scler 2004;10: 2-4. [ Links ]
78. Lepur D, Peterkovic V, Kalabric N. Neuromyelitis optica with CSF examination mimicking bacterial meningo-myelitis. Neurol Sci 2009;30:51-54. [ Links ]
79. Kira J. Multiple sclerosis in the Japanese population. Lancet Neurol 2003;2: 117-127. [ Links ]
80. Takahashi T, Miyazawa I, Misu T, Takano R, Naka-shima I, Fujihara K, et al. Intractable hiccup and nausea in neuromyelitis optica with anti-aguaporin-4 antibody: a herald of acute exacerbations. J Neurol Neurosurg Psichiatry 2008;79: 1075-1078. [ Links ]
81. Trojano M, Avolio C, Manzari C, Calo A, De Robertis F, Serio G, Livrea P. Multivariate analysis of predictive factors of multiple sclerosis course with a validated method to assess clinical events. F Neurosurg Psychiatry 1995;58:300-306. [ Links ]
82. Okinaka S, Tsubaki T, Kuroiwa Y, Toyokura Y, Imamura Y. Multiple sclerosis and allied disease in Japan; clinical characteristics. Neurology 1958; 8:756-763. [ Links ]
83. Kuroiwa Y, Igata A, Itahara K, Koshijima S, Tsubaki T. Nationwide survey of multiple sclerosis in Japan. Clinical analysis of 1084 cases. Neurology 1975;25:845-851. [ Links ]
84. Shibasaki H, McDonald WI, Kuroiwa Y. Racial modification of clinical picture of multiple sclerosis: comparison between British and Japanese patients. J Neurol Sci 1981;49:253-271. [ Links ]
85. Kira J. Western versus Asian types of multiple sclerosis: immunogenetically and clinically distinct disorders. Ann Neurol 1996;40:569-574. [ Links ]
86. Yamasaki K, Horiuchi I, Minohara M, Kawano Y, Ohyagi Y, Yamada T, Mihara F, Ito H, Nishimura Y, Kira J. HLA-DPB1*0501-associated opticospinal multiple sclerosis: clinical, neuroimaging and immunegenetic studies. Brain 1999;122:1689-1696. [ Links ]
87. Hung T P. Multiple sclerosis amongst Chinese in Taiwan. J Neurol Sci 1976;27:459-484. [ Links ]
88. Tabira T, et al. Neuropathological features of MS in Japan. In: Kuroiwa Y, Kurland LT, eds. Multiple Sclerosis East and West, Japan: Kyushu University Press 1982;273-295. [ Links ]
89. Ikuta F. Comparison of MS pathology between 70 American and 75 Japanese autopsy cases. Mult Scler 1982;297-306. [ Links ]
90. Barkhof F, Filippi M, Miller DH, Scheltens P, Campi A, Polman CH, et al. Comparison of MRI criteria at first presentation to predict conversion to clinically definite multiple sclerosis. Brain 1997;120:2059-2069. [ Links ]
91. Pittock SJ, Lennon VA, Krecke K, Wingerchuck DM, Lucchinetti C F, Weinshenker BG. Brain abnormalities in neuromyelitis optica. Arch Neurol 2006;63:390-396. [ Links ]
92. Kim W, Kim S, Lee SH, Li X F, Kim HJ. Brain abnormalities as an initial manifestation of neuromyelitis optica spectrum disorder. MSJ 2011:1-6. [ Links ]
93. Deodhar AA, Hochenedel T, Bennett RM. Longitudinal involvement of the spinal cord in a patient with lupus related transverse myelitis. J Rheumatol 1999;26:446-449. [ Links ]
94. Tellez-Zenteno J F, Remes-Troche JM, Negrete-Pulido RO, Dávila-Maldonado L. Longitudinal myelitis associated with systemic lupus Erythematosus: clinical features and magnetic resonance imaging of six cases. Lupus 2001;10:851-856. [ Links ]
95. Birnbaum J, Kerr D. Devic's syndrome in a woman with systemic lupus Erythematosus: diagnostic and therapeutic implications of testing for the neuromyelitis optica IgG autoantibody. Arthritis Care Res 2007;57:347-351. [ Links ]
96. Paira S, Benegas M, Ortiz A, Uña C, Rillo O, Mannucci P, Allievi A. Association of neuromyelitis optic (NMO) with autoimmune disorders: report of two cases and review of the literature. Clin Rheumatol 2010;29: 1335-1338. [ Links ]
97. McKeon A, Lennon VA, Lotze T, Tenenbaum S, Ness JM, Rensel M, et al. CNS aquaporin-4 autoimmunity in children. Neurology 2008;71:93-100. [ Links ]
98. Komolafe MA, Komolafe EO, Sunmonu TA, Olateju SO, Asaleye CM, Adesina OA, et al. New onset neuromyelitis optica in young Nigerian woman with possible antiphospholipid syndrome: a case report. J Med Case Reports 2008;2:348. [ Links ]
99. Ferreira S, Marques P, Carneiro E, D'Cruz D, Gama G. Devic's syndrome in systemic lupus erythematosus and probable antiphospholipid syndrome. Rheumatology (Oxford) 2005;44:693-695. [ Links ]
100. Chan AY, Liu DT. Devic's syndrome in systemic lupus erythematosus and probable antiphospholipid syndrome. Rheumatology (Oxford) 2006;45:120-121. [ Links ]
101. Mehta LR, Samuelson MK, Kleiner AK, Goodman AD, Anolik JH, Looney RJ, et al. Neuromyelitis optica spectrum disorder in a patient with systemic lupus erythematosus and anti-phospholipid antibody syndrome. Mult Scler 2008;14:425-427. [ Links ]
102. Lehnhardt FG, Impekoven P, Rubbert A, Burghaus L, Neveling M, Heiss WD, et al. Recurrent longitudinal myelitis as primary manifestation of SLE. Neurology 2004;63:1976. [ Links ]
103. Karussis D, Leker RR, Ashkenazi A, Abramsky O. A subgroup of unusual clinical manifestations: do they represent a new nosological entity? Ann Neurol 1998;44:629-634. [ Links ]
104. Hummers LK, Krishnan C, Casciola-Rosen L, Rosen A, Morris S, Mahoney JA, et al. Recurrent transverse myelitis associates with anti-Ro (SSA) autoantibodies. Neurology 2004;62:147-149. [ Links ]
105. Heinlein AC, Gertner E. Marked Inflammation in catastrophic longitudinal myelitis associated with systemic lupus erythematosus. Lupus 2007;16:823-826. [ Links ]
106. Birnbaum J, Kerr D. Optic neuritis and recurrent myelitis in a woman with systemic lupus erythematosus. Nat Clin Pract Rheumatol 2008;4:381-386. [ Links ]
107. Birnbaum J, Kerr D. Devic's syndrome in a woman with systemic lupus erythematosus: diagnostic and therapeutic implications of testing for the neuromyelitis optica IgG autoantibody. Arthritis Rheum 2007;57: 347-351. [ Links ]
108. Mochizuki A, Hyashi A, Hisahara S, Shoji S, et al. Steroid responsive Devic's variant in Sjögren's syndrome. Neurology 2000; 54:1391-1392. [ Links ]
109. Javed A, Balabanov R, Arnason BWG, Kelly TJ, Sweiss NJ, Pytel P, et al. Minor salivary gland inflammation in Devic's disease and longitudinally extensive myelitis. Mult Scler 2008;14(6):809-814. [ Links ]
110. Morgen K, McFarland H F, Pillemer SR. Central nervous system disease in primary Sjögren's syndrome: the role of magnetic resonance imaging. Semin Arthritis Rheum 2004;34(3):623-630. [ Links ]
111. Kira J, Kawano Y. Recurrent optic neuromyelitis with endocrinopathies. Neurology 1997;49:1475-1476. [ Links ]
112. Hui AC, Wong RS, Ma R, Kay R. Recurrent optic neuromyelitis with multiple endocrinopathies and autoimmune disorders. J Neurol 2002;249:784-785. [ Links ]
113. Petraviæ D, Habek M, Supe S, Brinar VV. Recurrent optic neuromyelitis with endocrinopathies: a new syndrome or a just coincidence? Mult Scler 2006;12:670-673. [ Links ]
114. Keegan M, Pineda AA, McClelland RL, Darby CH, Rodriguez M, Weinshenker BG.l. Plasma Exchange for severe attacks of CNS demyelination: predictors of response. Neurology 2002;58:143-146. [ Links ]
115. Theodoridou A, Settas L. Demyelination in rheumatic diseases. J Neurol Neurosurg Psychiatry 2006; 77:290-329. [ Links ]
116. Kaplin AI, Krishnan C, Deshpande DM, Pardo CA, Kerr DA. Diagnosis and management of acute myelopathies. Neurologist 2005;11:2-18. [ Links ]
117. Weinshenker BG, O'Brien PC, Petterson TM, Noseworthy JH, Lucchinetti C F, Dodick DW, et al. A randomized trial of plasma exchange in acute central nervous system inflammatory demyelinating disease. Ann Neurol 1999;46:878-886. [ Links ]
118. Llufriu S, Castillo J, Blanco Y, Ramió-Torrentà L, Río J, Vallès M, et al. Plasma Exchange for acute attacks of CNS demyelination: predictors of improvement at 6 months. Neurology 2009;73:949-953. [ Links ]
119. Watanabe S, Nakashima I, Misu T, Miyazawa I, Shiga Y, Fujihara K, et al. Therapeutic efficacy of plasma Exchange in NMO-IgG positive patients with neuromyelitis óptica. Mult Scler 2007;13:128-132. [ Links ]
120. Ruprecht K, Klinker E, Dintelmann T, Rieckmann P, Gold R. Plasma exchange for severe optic neuritis: treatment of 10 patients. Neurology 2004;63:1081-1083. [ Links ]
121. Bonnan M, Valentino R, Olindo S, Mehdaoui H, Smadja D, Cabre P. Plasma Exchange in severe spinal attacks associated with neuromyelitis optica spectrum disorder. Mult Scler 2009;15:487-492. [ Links ]
122. Miyamoto K, Kusunoki S. Intermittent plasmapheresis prevents recurrence in neuromyelitis optica. Ther Apher Dial 2009;13:505-508. [ Links ]
123. Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti C F, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology 2006;66:1485-1489. [ Links ]
124. Mandler RN, Ahmed W, Dencoff JE. Devic's neuromyelitis optica: a prospective study of seven patients treated with prednisone and azathioprine. Neurology 1998;51:1219-1220. [ Links ]
125. La Mantia L, Mascoli N, Milanese C. Azathioprine. Safety profile in multiple sclerosis patients. Neurol Sci 2007;28:299-303. [ Links ]
126. Killian JM, Bressler RB, Armstrong RM, Houston TX. Controlled pilot trial of monthly intravenous cyclophosphamide in multiple sclerosis. Arch Neurol 1988;45:27-30. [ Links ]
127. Weinstock-Guttman B, Ramanathan M, Lincoff N, Napoli SQ, Sharma J, Feichter J. Study of mitoxan-trone for the treatment of recurrent neuromyelitis optica (Devic Disease). Arch Neurol 2006;63:957-963. [ Links ]
128. Jacob A, Weinshenker BG, Violich I, McLinskey N, Krupp L, Fox RJ, et al. Treatment of neuromyelitis optica with rituximab: retrospective analysis of 25 patients. Arch Neurol 2008;65:1443-1448. [ Links ]
129. Kimby E. Tolerability and safety of rituximab (Mab-Thera). Cancer Treat Rev 2005;31:456-473. [ Links ]
130. Joseph FG, Scolding NJ. Neurolupus. Pract Neurol 2010;10:4-15. [ Links ]

