










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkRevista Colombiana de Reumatología
versão impressa ISSN 0121-8123
Rev.Colomb.Reumatol. v.18 n.4 Bogotá out./dez. 2011
PRESENTACIÓN DE CASOS Y REVISIÓN DE LA LITERATURA
1Internista-Reumatólogo. Profesor de la Universidad del Quindío.
2Médica y cirujana. Universidad Libre.
Correspondencia, Dr. Juan Pablo Restrepo: jprestrepo@lycos.com
Los autores declaran no presentar ningún conflicto de interés al momento de la redacción del manuscrito.
Recibido: 3 de agosto de 2011 Aceptado: 23 de septiembre de 2011
Resumen
La vasculopatía livedoide es una entidad rara y crónica que afecta predominantemente los miembros inferiores. La etiología de la enfermedad es desconocida, pero en la histopatología se observa trombosis en la dermis sin compromiso inflamatorio de la pared del vaso. Se manifiesta por lesiones petequiales y por ulceraciones que sanan con cicatrices hiperpigmentadas alternando con áreas blanquecinas. Se han empleado múltiples tratamientos hasta el momento, sin total éxito.
Palabras clave: síndrome antifosfolípido, livedo reticularis, fisiopatología, terapia.
Summary
Livedoid vasculopathy is a rare, chronic disease that affects predominantly the lower limbs. The etiology of the disease is unknown, but histopathology shows thrombosis in the dermis without inflammatory changes in the vessel wall. It is manifested by petechial lesions and ulcerations that heal with scarring hyperpigmentation alternating with white areas. Multiple treatments have been used so far, without total success.
Key words: antiphospholipid syndrome, reticularis livedo, pathophysiology, therapy.
Introducción
La vasculopatía livedoide (VL) es una entidad rara y poco estudiada. El hallazgo histo-patológico más importante de la enfermedad es la trombosis de los vasos de la dermis sin compromiso inflamatorio alrededor del vaso. Las manifestaciones clínicas son el resultado del infarto vascular. La VL se presenta aislada o en asociación a otras enfermedades autoinmunes como el síndrome antifosfolípido.
Se describe a continuación el caso de una mujer de veinticinco años con VL en presencia de anticoagulante lúpico como única manifestación trombótica de la enfermedad.
Caso clínico
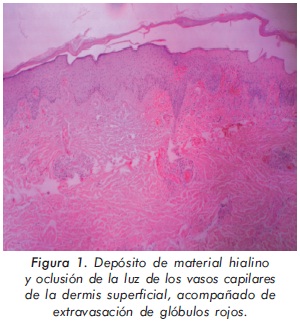
Mujer de veintiséis años con cuadro clínico de cinco años de evolución consistente en la aparición de lesiones violáceas en miembros inferiores que se oscurecen con el frío; al transcurrir el tiempo se le formaron pequeñas ulceraciones de carácter doloroso. No tiene antecedentes patológicos de importancia ni tampoco obstétricos. Al examen físico se encontró placas violáceas en piernas y pies, con pequeñas ulceraciones superficiales. Los laboratorios básicos se encontraban normales incluyendo los reactantes de fase aguda; las pruebas autoinmunes como ANAS, ENAS, anti-DNA, crioglobulinas, factor reumatoide y anticardiolipinas fueron normales. El anticoagulante lúpico por veneno de Russell fue reportado positivo en dos ocasiones con diferencia de doce semanas. Se realizó una biopsia de las lesiones cutáneas que reportó oclusión de capilares dérmicos sin componente inflamatorio en la pared del vaso sugiriendo una vasculopatía livedoide por síndrome antifosfolípido (Figura 1).
Se realizó manejo con ASA 100 mg al día acompañado de pentoxifilina 400 mg dos veces al día. A los tres meses de evolución las ulceraciones de los miembros inferiores habían sanado en su totalidad y sólo se observaba cicatrices producto de episodios previos sin tratamiento.
Definición
Esta enfermedad a lo largo de la historia ha tenido múltiples denominaciones. El término atrofia blanca en placa fue acuñado por Milian en 19291. La VL fue descrita en 1967 por Barel y Winkelm2. Otros nombres con los que se le conoce son livedo racemosa, vasculitis livedoide, vasculitis hialinizante segmentaria3,4, livedo reticularis con ulceración de verano y recientemente ha recibido el acrónimo de PURPLE (Painful purpuric Ulcers with Reticular Pattern of Lower Extremities)5.
Es una vasculopatía trombogénica de los capilares superficiales de las piernas, que puede ocasionar úlceras dolorosas y recurrentes que curan dejando una cicatriz atrófica y blanca, rodeada de telangiectasias y zonas de hiperpigmentación, de allí el nombre de atrofia blanca.
Epidemiología
Las mujeres son más afectadas que los hombres en una proporción de 4:1. Esta patología ocurre a cualquier edad pero su pico de incidencia es entre los 30 y 60 años6. Es una enfermedad crónica con exacerbaciones estacionales.
Patogénesis
La patogénesis de la enfermedad aún no es clara. Múltiples anomalías se han involucrado en la etiología de la VL (Tabla 1). Pizzo y colaboradores encontraron una liberación defectuosa del activador del plasminógeno tisular y Pittelkow demostró un incremento en los niveles del inhibidor del plasminógeno tisular. Tsutsu7 describió una disminución de la expresión de la trombomodulina la cual normalmente es antitrombótica y aumenta la activación de la proteína C. Otros hallazgos que sugieren un estado vasooclusivo son el déficit de proteína C8 y antitrombina III9, y el aumento de la agregación plaquetaria10. También se han encontrado niveles elevados de homocisteina en algunos pacientes con VL11. Hay dos formas de la enfermedad: una primaria o idiopática y otra secundaria. Los anticuerpos antifosfolípido pueden interactuar con células endoteliales y otros elementos celulares de los vasos sanguíneos llevando a vasoconstricción12. Estos anticuerpos van dirigidos contra fosfolípidos de la membrana de las células endoteliales, llevando a la producción de sustancias procoagulantes como el factor tisular, endotelina 1, inhibidor del activador del plasminógeno y finalmente se presenta la trombosis observada en el síndrome antifosfolípido (Tabla 1).

Los estados hipercoagulables y la enfermedad autoinmune son las dos condiciones más comúnmente asociadas con esta patología. La VL ha sido reportada en relación con síndrome antifosfolípido (SAF)13-15, lupus eritematoso sistémico (LES), esclerosis sistémica y arteriosclerosis16.
Histopatología
Se deben realizar varias biopsias tanto de las lesiones blanquecinas centrales como de las violáceas periféricas con el fin de aumentar el rendimiento diagnóstico17.
Los hallazgos en la patología varían dependiendo del estadio de la lesión. En las lesiones iniciales hay formación de trombos hialinos en la luz de los vasos de pequeño calibre de la dermis superficial. La presencia de trombosis capilar extensa obliga a descartar patología trombogénica18. Habitualmente hay necrosis de la dermis superficial con cambios en la epidermis tales como atrofia, paraqueratosis y ulceración de la misma. Puede haber infiltrado linfohistiocitario perivascular supeficial pero mínimo. También es común observar extravasación de glóbulos rojos.
En fases avanzadas hay engrosamiento, hialinización de los vasos sanguíneos con proliferación vascular. En las placas ulceradas se observa además de los cambios vasculares anteriormente mencionados, esclerosis dérmica, linfangiectasias con escasa inflamación de la epidermis.
La ausencia de leucocitoclasia alrededor de los vasos es el hallazgo distintivo más importante para diferenciarla de la vasculitis primaria19. La inmunofluorescencia directa usualmente muestra depósito de inmunoglobulinas, fibrina, complemento, pero son inespecíficos.
Manifestaciones clínicas
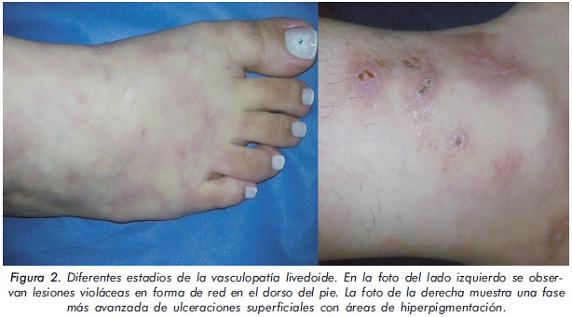
Se presentan unas lesiones reticulares o violáceas desproporcionadamente dolorosas alrededor de los maléolos conocidas como livedo racemosa; dichas lesiones pueden evolucionar a úlceras en sacabocado de 3-8 mm de diámetro (Figura 2). Las piernas son el lugar más frecuentemente afectado seguidas de los tobillos y la superficie dorsal del pie en menor proporción20. Posteriormente de días a semanas pueden aparecer úlceras rodeadas por telangiectasias puntiformes. La fase final de la enfermedad es llamada atrofia blanca debido a que estas lesiones pueden sanar con una cicatriz de menos de 1 cm de diámetro de color blanco porcelana.
Diagnóstico diferencial
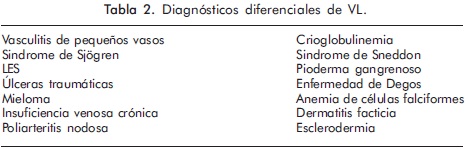
La lista de diagnóstico diferencial de la VL es extensa; por lo tanto, la historia clínica, el examen físico, las pruebas de laboratorio y la histopatología son los pilares en el diagnóstico de la enfermedad. Aspectos claves para el diagnóstico de la enfermedad son la evolución de las lesiones, la ubicación en miembros inferiores y la trombosis con ausencia de inflamación en la pared del vaso.
En la fase ulcerosa puede confundirse con vasculitis de pequeños vasos, pioderma gangrenoso, dermatitis facticia y enfermedad de Degos. La lesiones de atrofia blanca no son exclusivas de la VL pues pueden presentarse en esclerodermia, LES e insuficiencia venosa crónica21,22. En la Tabla 2 se encuentra el listado de los diagnósticos diferenciales en VL.
Tratamiento
El tratamiento de la VL idiopática es un reto. Como medidas generales se recomienda mantener los miembros inferiores elevados para ayudar a la cicatrización de las úlceras, así como evitar el consumo de cigarrillo.
Se ha empleado el ácido acetil salicílico, clopidogrel, activador del plasminógeno tisular, prostaciclina, beraprost, danazol, ketanserina, pentoxifilina y heparina de bajo peso molecular con resultados variables23-29. En caso de falta de respuesta a alguno de estos tratamientos se puede utilizar la combinación de ácido acetil salicílico y pentoxifilina30. En el caso de existir anticuerpos antifosfolípidos la anticoagulación con warfarina sería una alternativa razonable. Buenos resultados se han encontrado en pacientes con VL refractaria al tratamiento convencional con el uso de inmunoglobulina EV 4 gr/kgr en un periodo de seis semanas31. Zeni y colaboradores mostraron un caso de respuesta exitosa con rituximab en una mujer de treinta y ocho años quien había recibido tratamiento con anticoagulación e inmunosupresión32.
Discusión
El SAF fue descrito por primera vez por Hughes y colaboradores y se ha definido como una trombofilia adquirida por la presencia de anticuerpos dirigidos contra los fosfolípidos de membranas y/o sus cofactores. De acuerdo con los criterios de clasificación de Sidney se define SAF como la ocurrencia de uno o más episodios de trombosis de pequeños vasos arterial o venosa de cualquier órgano o tejido o morbilidad durante el embarazo, descrita como una muerte fetal no explicada más allá de la semana 10, uno o más nacimientos prematuros normales debido a preeclampsia o eclampsia severa, tres o más abortos consecutivos antes de la semana 10 de gestación sin alteraciones cromosómicas, acompañados de anticardiolipinas en títulos medios o altos o anticoagulante lúpico presentes en dos ocasiones con un intervalo de doce semanas33. Una amplia variedad de manifestaciones cutáneas han sido descritas asociadas con el SAF, las cuales pueden ser el primer signo de la enfermedad. Éstas incluyen livedo reticularis, tromboflebitis, púrpura, hematomas, nódulos en la piel, sangrado periungueal, acrocianosis, gangrena digital, dermografismo y VL, entre otras34. En una serie de 200 pacientes con SAF, se encontró un 49% de manifestaciones cutáneas35. La VL es una entidad rara cuyo mecanismo fisiopatológico parece involucrar un trastorno de la coagulación o de la fibrinólisis que conduce a oclusión de pequeños vasos de la dermis. El tratamiento de la VL es difícil. La mayoría de información disponible está basada en gran medida en informes de casos y series de casos.
Se describe el caso de una mujer de 26 años sin patología obstétrica reconocida que presentó un compromiso ulceroso en miembros inferiores sin compromiso vascular sistémico, encontrándose en la biopsia de piel algunas zonas de trombosis dérmicas sin afectación inflamatoria del vaso. No se pudo objetivar mediante paraclínicos de otra posible etiología. Ajustados a la clasificación de Sydney, la paciente tenía documentación de trombosis, en este caso histopatológica, sumada a criterios de laboratorio como el anticoagulante lúpico en dos ocasiones con doce semanas de diferencia, con lo cual se realizó un diagnóstico de VL asociada a SAF. La afección cutánea de SAF como una manifestación de la enfermedad ha sido descrita en la literatura. En este reporte se realizó un tratamiento basado en el uso de un antiagregante plaquetario acompañado de un agente hemorreológico con buenos resultados; es de resaltar que en la mayoría de casos se requiere de otros tratamientos para controlar la enfermedad.
Referencias
1. Hairston B, Davis M, Pittelkow M, Ahmed I. Livedoid vasculopathy: further evidence for procoagulant pathogenesis. Arch Dermatol 2006;142:1413-1418. [ Links ]
2. Bard J, Winkelmann R. Livedo vasculitis. Segmental hyalinizing vasculitis of the dermis. Arch Dermatol 1967;96:489-499. [ Links ]
3. Milian G. Les atrophies cutanées syphilitiques. Bull Soc Franc Derm Syph 1929;36:865-871. [ Links ]
4. Winkelmann R, Schroeter A, Kierland R, Ryan T. Clinical studies of livedoid vasculitis: segmental hyalinizing vasculitis. Mayo Clin Proc 1974;49:746-750. [ Links ]
5. Papi M, Didona B, De Pita O, Silvestri L, Ferranti G, Gantcheva M, Chinni L. PURPLE (atrophie blanche): Clinical, histological and immunological study of twelve patients. J Eur Acad Dermatol Venereol 1997;9:129-133. [ Links ]
6. Maessen-Visch M, Koedam M, Hamulyák K, Neumann H. Atrophie blanche. Int J Dermatol 1999; 38:161-172. [ Links ]
7. Tsutsui K, Shirasaki F, Takata M, Takehara K. Successful treatment of livedo vasculitis with beraprost sodium: a possible mechanism of thrombomodulin upregulation. Dermatol 1996;192:120-124. [ Links ]
8. Baccard M, Vignon-Pennamen M, Janier M, Scrobohaci M, Dubertret L. Livedo vasculitis with protein C system deficiency. Arch Dermatol 1992; 128:1410-1411. [ Links ]
9. Hegemann B, Helmbold P, Marsch W. Livedoid vasculitis with ulcerations: the role of antithrombin III deficiency and its therapeutic consequences. Arch Dermatol 2002;138:841-842. [ Links ]
10. Drucker C, Duncan W. Antiplatelet therapy in atrophie blanche and livedo vasculitis. J Am Acad Dermatol 1982;7:359-363. [ Links ]
11. Meiss F, Marsch W, Fischer M. Livedoid vasculopathy. The role of hyperhomocysteinemia and its simple therapeutic consequences. Eur J Dermatol 2006; 16:159-162. [ Links ]
12. Uthman I, Khamashta M. Livedo racemosa: a striking dermatological sign for the antiphospholipid syndrome. J Rheumatol 2006;33:2379-2382. [ Links ]
13. Grattan C, Burton J, Boon A. Sneddon's syndrome (livedo reticularis and cerebral thrombosis) with livedo vasculitis and anticardiolipin antibodies. Br J Dermatol 1989;120:441-447. [ Links ]
14. Acland K, Darvay A, Wakelin S, Russell-Jones R. Livedoid vasculitis: a manifestation of the antiphos-pholipid syndrome? Br J Dermatol 1999;140:131-135. [ Links ]
15. Grob J, Bonerandi J. Thrombotic skin disease as a marker of the anticardiolipin syndrome. Livedo vasculitis and distal gangrene associated with abnormal serum antiphospholipid activity. J Am Acad Dermatol 1989;20:1063-1069. [ Links ]
16. Leonard A, Pomeranz M, Franks A Jr. A case of livedoid vasculopathy in a 22-year-old man. J Drugs Dermatol 2004;3:678-679. [ Links ]
17. In S, Han J, Kang H, Lee E, Kim Y. The histopathological characteristics of livedo reticularis. J Cutan Pathol 2009;36:1275-1278. [ Links ]
18. Shimizu A, Tamura A, Yamanaka M, Amano H, Nagai Y, Ishikawa O. Case of livedoid vasculopathy with extensive dermal capillary thrombi. J Dermatol 2010;37:94-97. [ Links ]
19. Gibson L, Su W. Cutaneous vasculitis. Rheum Dis Clin North Am 1995;21:1097-1113. [ Links ]
20. Weinstein S, Piette W. Cutaneous manifestations of antiphospholipid antibody syndrome. Hematol Oncol Clin North Am 2008;22:67-77. [ Links ]
21. Criado P, Rivitti E, Sotto M, de Carvalho J. Livedoid vasculopathy as a coagulation disorder. Autoimmun Rev 2011;10:353-360. [ Links ]
22. Goerge T. Livedovaskulopathie. Pathogenese, Diagnostik und Therapie des Hautinfarkts. Hautarzt 2011;62:627-636. [ Links ]
23. Klein K, Pittelkow M. Tissue plasminogen activator for treatment of livedoid vasculitis. Mayo Clin Proc 1992;67:923-933. [ Links ]
24. Hoogenberg K, Tupker R, van Essen L, Smith A, Kallenberg C. Successful treatment of ulcerating livedo reticularis with infusions of prostacyclin. Br J Dermatol 1992;127:64-66. [ Links ]
25. Yamamoto M, Danno K, Shio H, Imamura S. Antithrombotic treatment in livedo vasculitis. J Am Acad Dermatol 1988;18:57-62. [ Links ]
26. Rustin M, Bunker C, Dowd P. Chronic leg ulceration with livedoid vasculitis, and response to oral ketanserin. Br J Dermatol 1989;120:101-105. [ Links ]
27. Hsiao G, Chiu H. Low-dose danazol in the treatment of livedoid vasculitis. Dermatol 1997;194:251-255. [ Links ]
28. Hsiao G, Chiu H. Livedoid vasculitis. Response to low-dose danazol. Arch Dermatol 1996;132:749-751. [ Links ]
29. Hairston B, Davis M, Gibson L, Drage L. Treatment of livedoid vasculopathy with low-molecular-weight heparin: report of 2 cases. Arch Dermatol 2003;139: 987-990. [ Links ]
30. Callen J. Livedoid vasculopathy: what it is and how the patient should be evaluated and treated. Arch Dermatol 2006;142:1481-1482. [ Links ]
31. Pitarch G, Rodríguez-Serna M, Torrijos A, Oliver V, Fortea J. Treatment of livedoid vasculopathy with short-cycle intravenous immunoglobulins. Acta Derm Venereol 2005;85:374-375. [ Links ]
32. Zeni P, Finger E, Scheinberg M. Successful use of rituximab in a patient with recalcitrant livedoid vasculopathy. Ann Rheum Dis 2008;67:1055-1056. [ Links ]
33. Miyaki S, Lockshin M, Atsumi T, Branch D, Brey R, Cervera R et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 2006;295-306. [ Links ]
34. Rai R, Sekar C, Kumaresan M. Antiphospholipid syndrome in dermatology: An update. Indian J Dermatol Venereol Leprol 2010;76:116-124. [ Links ]
35. Asherson R, Francés C, Iaccarino L, Khamashta M, Malacarne F, Piette J et al. The antiphospholipid antibody syndrome: diagnosis, skin manifestations and current therapy. Clin Exp Rheumatol 2006; 24:46-51. [ Links ]

