










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Reumatología
Print version ISSN 0121-8123
Rev.Colomb.Reumatol. vol.19 no.2 Bogotá Apr./June 2012
REPORTE DE CASO
Síndrome pulmón riñón en vasculitis sistémica por inmunoglobulina A secundaria a linfoma
Lung-kidney sindrome in systemic vasculitis by inmunoglobulin A due to lymphoma
David Andrés Ocampo Ramírez1, César A Restrepo Valencia1
1Sección de Nefrología. Departamento de Medicina Interna. Hospital Militar Central. Universidad de Caldas. Manizales, Colombia.
Correspondencia: César A. Restrepo: caugustorv@une.net.co
Los autores declaran no presentar ningún conflicto de interés al momento de la redacción del manuscrito.
Recibido: 10 de enero de 2012. Aceptado: 7 de mayo de 2012.
Resumen
Paciente de 35 años de edad, quien debuta con síndrome de pulmón riñón secundario a vasculitis pulmonar y depósitos renales granulosos de IgA, en quien se diagnostica vasculitis sistémica por IgA, posteriormente, presenta compromiso pancreático y cutáneo (Púrpura de Henoch - Schönlein), falleciendo ocho meses más tarde por linfoma difuso de células B de alto grado. Este es el reporte de caso número catorce en el que se establece asociación entre neoplasia hematológica y Púrpura de Henoch - Schönlein.
Palabras clave: Púrpura de Henoch - Schönlein, síndrome pulmón riñón, linfoma difuso de células B de alto grado.
Summary
Patient was 35-years-old who presents with kidney-lung syndrome secondary to pulmonary vasculitis and renal granular deposits of IgA, in whom systemic vasculitis IgA was diagnosed, and then he had cutaneous and pancreatic involvement (Henoch Schönlein Purpura). He died eight months later by diffuse high grade B-cell lymphoma. This is the number fourteen case report of an association between hematological malignancy and Henoch - Schönlein.
Key words: Henoch - Schönlein Purpura, kidney-lung syndrome, diffuse high grade B-cell lymphoma.
Introducción
La hemorragia alveolar difusa (HAD) es una condición poco frecuente en la cual la sangre inunda los alvéolos1. Si la HAD se acompaña de alteraciones en la función renal, generalmente del tipo de glomerulonefritis rápidamente progresiva, se configura un síndrome pulmón riñón2. Las vasculitis son definidas como inflamación de los vasos sanguíneos y originan el 60-70% de los casos reportados de Síndrome Pulmón Riñón (SPR); el calibre y tamaño de los vasos son los aspectos más importantes para la clasificación3. La púrpura de Henoch-Schönlein (PHS) es una vasculitis de pequeños vasos no granulomatosa, ANCA negativa, mediada por IgA, y sus criterios diagnósticos actuales fueron establecidos en el año 2005 por la Sociedad Europea de Reumatología Pediátrica, y reafirmados por el Colegio Americano de Reumatología y Sociedad Europea de Nefrología Pediátrica en el año 2006, e incluyen púrpura palpable (sin trombocitopenia ni coagulopatía), en presencia de por lo menos uno de los siguientes hallazgos: dolor abdominal difuso, artritis o artralgias, cualquier biopsia en la que predominaran los depósitos de IgA y compromiso renal (manifestado por hematuria o proteinuria)4. En adultos la PHS tiene curso y pronóstico diferentes al de los niños, y en varios casos se ha asociado a malignidad que incluye neoplasia hematológica. Se presenta el caso de una paciente con PHS que debuta como SPR, siendo posteriormente identificado secundario a linfoma difuso de células B de alto grado.
Presentación del caso
Paciente de sexo femenino de 35 años de edad, procedente del área urbana del departamento de Caldas, sin antecedentes personales de importancia, quien consultó por cuadro clínico de veinte días de evolución caracterizado por edema en miembros inferiores y disnea progresiva, que avanzó a falla respiratoria, requiriendo traslado a unidad de cuidado intensivo para soporte ventilatorio.
Se realizó tomografía de tórax en la cual se evidenció imagen en vidrio esmerilado de predominio en dos tercios inferiores asociado a derrame pleural (Figura 1). En los laboratorios iniciales se reportó creatinina en 1.5 mg/dL; hemograma con leucocitos 7300 x mm3, neutrófilos 6600 x mm3, hemoglobina 12 gr/dL y plaquetas 140.000 x mm3; examen de orina con leucocituria, hematuria, cilindros granulosos y proteínas 30 mg/dL.
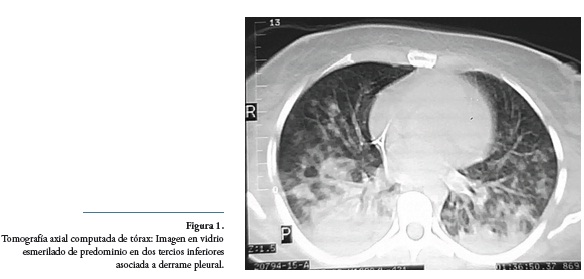
Se consideró como posibilidad etiológica del cuadro patología infecciosa o SPR por enfermedad del colágeno, se inició tratamiento con ampicilina sulbactam y amikacina, se realizó fibrobroncoscopia con lavado broncoalveolar, el cual no reportó sangrado y se tomaron muestras para estudio citológico y cultivo cuyo reporte fue negativo para aislamiento de bacterias, micobacterias y hongos.
Siete días más tarde la hemoglobina disminuyó a 8.6 gr/dL, sin evidencias de sangrado externo ni gastrointestinal y con bilirrubinas en rangos normales. Sin presentar mejoría clínica ni radiológica y con diagnóstico de SPR se inició pulso de esteroide con metilprednisolona 500 mg intravenoso (IV) diario, por tres días, continuándose con prednisolona 60 mg al día asociada a ciclofosfamida 50 mg, vía oral, por día.
Por consideración de injuria pulmonar aguda en cuadro clínico de sepsis, con pobre respuesta al manejo antibiótico y a la espera de la valoración por nefrología, se optó por practicar inicialmente biopsia pulmonar a cielo abierto, cuyo reporte fue: “pérdida de la arquitectura normal con compromiso centrado en vasos y caracterizado por infiltrado inflamatorio de neutrófilos y linfocitos que comprometen la pared de capilares y arterias de mediano calibre, capilaritis y vasculitis, con presencia de microtrombos en algunos de ellos, hemorragia alveolar, reciente y antigua, dada por hematíes y escasos hemosiderófagos. Diagnóstico: vasculitis con hemorragia pulmonar”. No se practicó inmunofluorescencia a la muestra.
Posteriormente, nefrología evaluó a la paciente, y con impresión diagnóstica de síndrome nefrítico en el contexto de un SPR, recomendó realizar biopsia renal, la cual se practicó en forma percutánea, siendo su reporte: “engrosamiento irregular y focal de membrana basal con adherencias a cápsula de Bowman, ensanchamiento del mesangio y proliferación mesangial difusa; en la inmunofluorescencia depósitos granulosos en mesangio y membrana basal, predominantemente de IgA y escasos de IgM; negativo para IgG, C1q, C3, C4, fibrinógeno, cadenas kappa y lambda”.
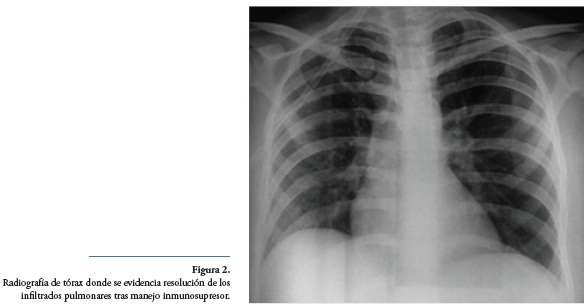
La paciente evolucionó favorablemente con resolución de los infiltrados alveolares, y sin evidencia de patología pulmonar que ocupara espacio (Figura 2). Fue dada de alta hospitalaria dos semanas más tarde, en tratamiento con prednisolona y ciclofosfamida oral.
Un mes después consultó por cuadro de tres días de evolución de dolor abdominal localizado en epigastrio y mesogastrio, tipo cólico y punzante. Se practicaron ecografía abdominal total y colonoscopia, con resultados normales. La endoscopia digestiva alta reportó gastropatía crónica antral, y en la tomografía de abdomen se hallaron 2 imágenes sugestivas, de pequeños abscesos en cabeza de páncreas, de aproximadamente 2x2 cm y probable absceso peripancreático de aproximadamente 4x3 cm (Figura 3). En cortes altos de abdomen no se observó compromiso de las bases pulmonares; las pruebas de función pancreática con lipasa 1021 U/L y amilasa 312 U/L fueron elevadas.
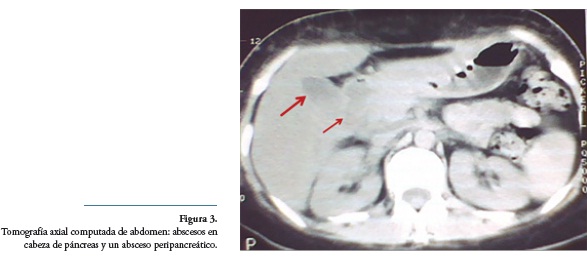
Se sospechó cuadro de vasculitis pancreática o un cuadro neoplásico, se suspendió ciclofosfamida por no haber certeza de la etiología y se realizó colangiopancreatografía retrógrada endoscópica terapéutica, pero los quistes no estaban comunicados con el conducto pancreático principal y no fue posible su drenaje, se decidió manejo médico con seguimiento imaginológico ambulatorio; tras obtener resolución del dolor abdominal fue dada nuevamente de alta en manejo con prednisolona.
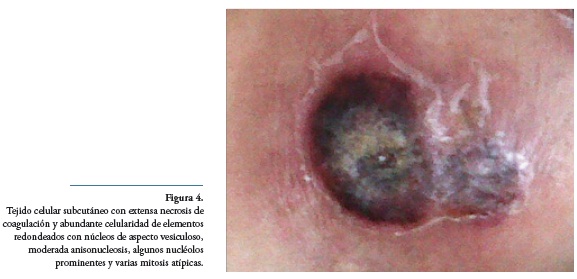
Tres meses después, presentó múltiples lesiones inicialmente de aspecto purpúrico palpable en glúteos y miembros inferiores, las cuales se tornaron nodulares y posteriormente ulcerativas, de 3x3 cm, con calor y edema local (Figura 4). La biopsia de éstas reportó “tejido celular subcutáneo con extensa necrosis de coagulación y con abundante celularidad representada por elementos redondeados con núcleos de aspecto vesiculoso, con moderada anisonucleosis, algunos nucléolos prominentes y varias mitosis atípicas”. Se consideró vasculitis sin respuesta a esteroides y se inició manejo con azatioprina y cloroquina.
Dos semanas más tarde inició con tos seca asociada a disnea intermitente y dolor localizado en hipocondrio izquierdo, progresando a falla respiratoria, en tomografía de tórax se observaron múltiples masas intraparenquimatosas con densidad de tejidos blandos, con captación heterogénea del medio de contraste, las cuales por sus características sugirieron lesiones de tipo metastásico (Figura 5).
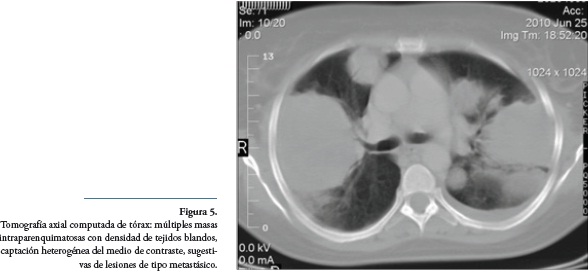
Se realizó nueva biopsia pulmonar hallándose “neoplasia maligna constituida por elementos celulares redondeados con marcado pleomorfismo y anisocariosis. Los núcleos presentaban formas ovoides en su mayoría, algunos eran núcleos vesiculosos y otros hipercromáticos. Los citoplasmas eran escasos y algunos de tinción eosinófila. Se encontraron varias mitosis atípicas. Se observó, además, abundante tejido necrótico. Inmunohistoquímica CK (-), S100 (-), vimentina (+), CD 45 (+), CD 3 (-), CD 15 (-), CD 20 (+), CD 30 (-), compatible con linfoma difuso de células B de alto grado”.
Finalmente, la paciente falleció dado su compromiso respiratorio y antes de iniciar terapia para el linfoma.
Discusión
La hemorragia alveolar difusa (HAD) es una condición poco frecuente que se puede clasificar de acuerdo con dos patrones de ocurrencia1 (Tabla 1):

- Asociada a vasculitis o capilaritis, con evidencia de neutrófilos que infiltran los vasos septales interalveolares y peribronquiales ocasionando la ruptura anatómica de los capilares y extravasación de células hemáticas dentro del alvéolo e intersticio, con producción de fibrina, exudado inflamatorio y necrosis fibrinoide.
- Hemorragia alveolar blanda sin capilaritis ni vasculitis cuando los glóbulos rojos se filtran al alvéolo sin evidencia de inflamación o destrucción de capilares alveolares, vénulas, o arteriolas.
Las características clínicas dependen de la enfermedad de base. La disnea, tos y fiebre son, generalmente, los síntomas iniciales que se presentan con mayor frecuencia de forma aguda o subaguda. Algunos pacientes manifiestan falla respiratoria. La hemoptisis puede estar ausente hasta en un tercio de los pacientes, debido a que el gran tamaño pulmonar le permite absorber grandes cantidades de sangre. El examen físico general es inespecífico, puede reflejar enfermedad del colágeno o vasculitis con púrpura palpable, lesiones oculares y hepatoesplenomegalia.1
Se debe sospechar el diagnóstico de HAD en presencia de infiltrados pulmonares bilaterales y caída en los niveles de hemoglobina; en todos los casos se debe utilizar antibiótico de amplio espectro hasta cuando el cuadro se haya aclarado2.
El lavado broncoalveolar muestra numerosos eritrocitos y siderófagos (macrófagos cargados de hemosiderina), permite excluir sitios de sangrado en vía aérea superior y descartar cuadro infeccioso. La biopsia pulmonar a cielo abierto y de otros órganos involucrados es necesaria para establecer la etiología. La biopsia transbronquial es poco útil debido al pequeño tamaño de los especímenes. La apariencia histológica de la hemorragia alveolar difusa es relativamente uniforme, cualquiera sea la causa1.
En muchas ocasiones la HAD se acompaña de daño renal conjunto que se conoce como SPR. Cuando la alteración renal es una glomerulonefritis rápidamente progresiva se configura un síndrome de Goodpasture, cuando se demuestra que es causada por anticuerpos anti-membrana basal glomerular, contra cualquier componente de esta membrana, se denomina enfermedad Anti-Membrana Basal Glomerular, pero si el antígeno hacia el cual está dirigida la respuesta inmune es el de Goodpasture se acepta el nombre de enfermedad de Goodpasture. La biopsia renal es imprescindible para determinar el diagnóstico y la severidad del compromiso glomerular5.
Las vasculitis sistémicas, enfermedad de Goodpasture y la enfermedad por anticuerpos antimembrana basal glomerular, son las entidades que con mayor frecuencia originan el SPR. En nuestra paciente la presencia de vasculitis (capilaritis) pulmonar y compromiso renal, con depósitos principalmente de inmunoglobulina A, limita las posibilidades diagnósticas a síndrome de Goodpasture, nefropatía por IgA y púrpura de Henoch-Schönlein.
En la enfermedad de Goodpasture el blanco principal para los anticuerpos antimembrana basal es el dominio carboxi-terminal no colágeno (NC1), de la cadena alfa 3, del colágeno tipo 4 (antígeno de Good-pasture). En la biopsia renal se detectan depósitos lineares, principalmente, de IgG sobre la membrana basal, pero puede presentarse por depósitos lineales de IgM e IgA, y raramente hay sólo depósitos de IgA6,7.
Reconocer el patrón de depósito de los anticuerpos es fundamental para un adecuado diagnóstico de los pacientes; en el caso actual por inmunofluorescencia el patrón fue de tipo granuloso lo cual descartó el síndrome de Goodpasture y nos centró en las dos patologías restantes.
La nefropatía por IgA o enfermedad de Berger, se caracteriza por depósitos granulares con predominio de IgA en el mesangio. Es más común en adolescentes y se caracteriza por episodios recurrentes de hematuria macroscópica concomitantes a faringitis o infección del tracto respiratorio superior, usualmente la función renal es normal y casi siempre C3 y C4 están normales5.
La nefropatía por IgA puede comprometer otros órganos además de los riñones; la capilaritis pulmonar ha sido reportada en solo 11 casos8. Presenta características histopatológicas y clínicas, incluyendo dolor abdominal y compromiso articular, similares a la PHS y puede encontrarse en pacientes que desarrollan púrpura posteriormente. A nivel gastrointestinal se ha descrito enteropatía por IgA en ausencia de lesiones cutáneas, de ahí que algunos autores sugieren que la nefropatía por IgA es una PHS sin rash9.
La Púrpura de Henoch - Schönlein se define como vasculitis leucocitoclástica sistémica que involucra arteriolas y vénulas, más comúnmente de la piel, glomérulos y tracto gastrointestinal10. Ocurre generalmente entre los 4 y 16 años de edad, con incidencia de 20 casos por 100.000 habitantes11, pero se han reportado casos de pacientes de 6 meses hasta 86 años de edad, por lo cual los criterios para su diagnóstico en el 2006 excluyeron el de la edad menor a 20 años. Las características clínicas pueden ser atípicas en los extremos de la vida y en adultos es, usualmente, una complicación más seria12,13.
Todos los pacientes con cuadro extra abdominal desarrollan púrpura y entre 57-69% está presente como manifestación inicial; el depósito de IgA cutáneo puede dar lugar clínicamente a la presencia de púrpura retiforme, poco descrita por los clínicos14.
El compromiso renal ocurre en 32-54% de los pacientes y de 75-85% lo desarrolla en las cuatro primeras semanas desde el compromiso cutáneo, aunque puede abarcar hasta los tres primeros meses, usualmente se manifiesta como hematuria, siendo la proteinuria sin hematuria poco común9,15. En adultos la prevalencia alcanza hasta 85% de los pacientes según la definición dada en diferentes series16. Al comparar con la nefropatía por IgA la biopsia renal es indistinguible entre ambas entidades, aunque la púrpura tiende a generar más depósitos inmunes en las paredes de las asas capilares (membrana basal glomerular) y compromiso extracapilar con formación de medias lunas.
En general la PHS puede comprometer cualquier órgano del aparato gastrointestinal9. Se puede manifestar con cólico localizado en las regiones periumbilical y epigástrica, que empeora luego de la ingesta en forma similar a angina o isquemia intestinal, secundario a vasculitis de pequeño vaso que genera necrosis de las vellosidades intestinales. El dolor abdominal puede ser signo de presentación en 12-19%, siendo severo hasta en 42% de los pacientes y es concomitante con náuseas, emesis o sangrado. Puede haber distensión e imitar abdomen agudo que resulta, en ocasiones, en cirugías innecesarias. Las principales indicaciones quirúrgicas son intususcepción, perforación e infarto.9
Se reporta la vasculitis del páncreas por PHS como una rara complicación, descrita inicialmente en 1965 y hasta la fecha con 14 casos documentados. El dolor abdominal puede preceder a la púrpura en 14-36% de los casos17.
Debido a que la PHS es mucho más rara en adultos que en niños los criterios no son igualmente aplicables dado que la clínica puede variar; en un estudio retrospectivo de 250 pacientes adultos con seguimiento promedio de 14,8 años se encontró que el 96% de los pacientes presentó púrpura palpable, y siempre localizada en los miembros inferiores, en el 83% de los casos fue la manifestación inicial y compromiso de cara y mucosas sólo se detectó en el 5% de los pacientes. Vesículas y lesiones cutáneas necróticas semejantes a las de nuestra paciente ocurrieron en el 35% de los pacientes16.
Por otra parte, la presentación de vasculitis cutánea o sistémica difiere en el adulto, ya que se estima que de 2.5 a 5% son secundarias a neoplasias, presentándose hasta en un 71% de los casos de forma concomitante con el tumor o recaídas en el mismo18. En un estudio francés, de 10 años de seguimiento, que incluyó 60 pacientes, se encontraron lesiones cutáneas frecuentes e inespecíficas tipo pápulas, nódulos, bulas, ulceraciones y lesiones necróticas; éstas pueden aparecer inclusive hasta 25 meses antes de la aparición del tumor19.
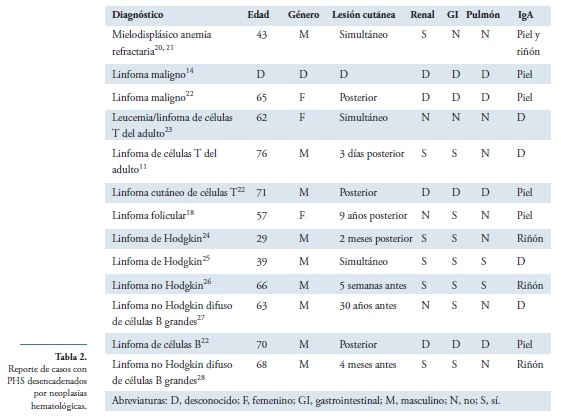
A la fecha hay reporte de trece casos, en la literatura, en los cuales la PHS es desencadenada por neoplasia hematológica, sin incluir los asociados a policitemia vera y trombocitemia esencial como se muestra en la tabla 211,14,18,20-28.
Los pacientes con neoplasias de células T pueden manifestar lesiones cutáneas específicas o no del tumor. La presencia de nódulos se explica por la infiltración y proliferación de las células tumorales en la piel; el origen de otros tipos de lesión es incierto, a pesar que usualmente se relacionan con la respuesta del huésped entre otros factores. Las lesiones purpúricas diseminadas son muy raras y en ocasiones son secundarias a trombocitopenia; otros mecanismos causales son aún materia de controversia. Histológicamente los pequeños vasos también pueden ser infiltrados causando trombosis sin hallazgo de vasculitis22, 23, 24.
En cuanto a la fisiopatología de la PHS asociada a enfermedad neoplásica se plantean de forma especulativa varias hipótesis: producción de IgA-2 por la mucosa que se deposita en sitios distantes; antígenos tumorales que pueden llevar a la producción de IgA-1 por las células B; anormalidades en el sistema inmune que inducen a que los linfocitos que promueven IgM lo hagan hacia IgA. Una vez la inmunoglobulina se deposita en los tejidos el disparador de la vasculitis es incierto, pero podrían intervenir Fc, complemento y procesos inmunes mediados por las células T. Otros autores proponen la actuación de las células tumorales como neoantígenos con similitud molecular a antígenos de las paredes de los vasos sanguíneos, disminución de la depuración de complejos inmunes y producción aberrante de citoquinas inflamatorias18, 29.
En relación con la terapia, los hallazgos son controversiales, un reporte de caso encontró que el tratamiento del tumor resultó en resolución de la púrpura;24 por otra parte, se ha encontrado que el tratamiento de la neoplasia también se ha asociado con la aparición de PHS que se cree secundaria a la destrucción de células tumorales o cambios en los antígenos de superficie18, 29.
Finalmente, se evidencia alteración de la función supresora con imbalance de linfocitos Th1 y Th2; producción en exceso de anticuerpos IgA por parte de los linfocitos B hiperreactivos, que puede crear un medio conductivo para el desarrollo de una reacción por complejos inmunes y generación de IgA contra la mucosa y el endotelio secundario a citoquinas como la CK1814.
En nuestro caso se presenta una paciente con púrpura de Henoch Schönlein que de forma concomitante presenta linfoma difuso de células B, cuyo reporte sería el número catorce en la literatura, de posible asociación a neoplasia hematológica; consideramos que la paciente cursó con PHS, la cual, por tratarse de un adulto, tuvo una presentación atípica con necrosis. Es evidente que hay hallazgos que demuestran que pueden presentarse lesiones cutáneas posteriormente a la alteración renal cuando se trata de neoplasias, desconocemos el porqué dicho hallazgo clínico es diferente cuando la etiología de la púrpura es benigna, pero tal vez es debido a injuria más persistente al permanecer la alteración de base. En algunas ocasiones, según lo mostrado en la literatura, en pacientes con PHS que son adultos y cuya causa no sea evidente pudiera buscarse relación con neoplasias.
Referencias
1. Ioachimescu O C, Stoller J K. Diffuse alveolar hemorrhage: Diagnosing it and finding the cause. Cleve Clin J Medi 2008; 75:258-280. [ Links ]
2. Papiris S A, Manali E D, Kalomenidis I, Kapotsis G E, Karakatsani A, Roussos C. Bench-to-bedside review: Pulmonary-renal syndromes and update for the intensivist. Critical Care 2007; 11:213-223. [ Links ]
3. Sunderkötter C, Sindrilaru A. Clinical classification of vasculitis. Eur J Dermatol 2006; 16:114-124. [ Links ]
4. Ozen S, Ruperto N, Dillon M J, Bagga A, Barron K, Davin J C, Kawasaki T, Lindsley C, Petty R E, Prieur A M, Ravelli A, Woo P. EULAR/PReS endorsed consensus criteria for the classification of childhood vasculitides. Ann Rheum Dis 2006; 65:936-941. [ Links ]
5. Lau K K, Wyatt R J. Glomerulonephritis. Adolesc Med clinics 2005; 16:67-85. [ Links ]
6. Lionaki S, Jennete J C, Falk R J. Anti-neutrophil cytoplasmic (ANCA) and anti-glomerular basement membrane (GBM) autoantibodies in necrotizing and crescentic glomerulonephritis. Semin Immunopathol 2007; 29:459-474. [ Links ]
7. Restrepo C A. Goodpasture's Syndrome due to IgA in a patient with clinical diagnosis of Henoch Schonlein's purpura. Acta Méd Colomb 2005; 30:285-288. [ Links ]
8. Srivaths P, Dishop M K, Elidemir O, Eldin K, Browne L, Elenberg E. IgA nephropathy presenting with renal failure and pulmonary hemorrhage. Pediatr Nephrol 2010; 25:535-538. [ Links ]
9. Ebert E. C. Gastrointestinal Manifestations of Henoch-Schonlein Purpura. Dig Dis Sci 2008; 53:2011-2019. [ Links ]
10. Mills J A, Michel B A, Bloch D A, et al. The American College of Rheumatology, 1990 criteria for the classification of Henoch-Schonlein purpura. Arthritis Rheum 1990; 33:1114-1121. [ Links ]
11. Tabata R, Tabata C, Namiuchi S, Terada M, Yasumizu R, Okamoto T, Nagai T. Adult T-cell lymphoma mimicking Henoch-Schönlein purpura. Mod Rheumatol 2007; 17:57-62. [ Links ]
12. Saulsbury F T. Henoch-Schönlein purpura. Curr Opin Rheumatol 2001; 13:35-40. [ Links ]
13. Farley T A, Gillespie S, Rasoulpour M, Tolentino N, Hadler J L, Hurwitz E. Epidemiology of a Cluster of Henoch-Schonlein Purpura. Am J Dis Child 1989; 143:798-803. [ Links ]
14. Cynthia M M, Neil C A. A Clinical and Histologic Study of 37 Cases of Immunoglobulin A-Associated Vasculitis. Am J Dermatopathol 1999; 1:234-240. [ Links ]
15. Chartapisak W, Opastiraku S L, Willis N S, et al. Prevention and treatment of renal disease in Henoch-Schonlein purpura: a systematic review. Arch Dis Child 2009; 94:132-137. [ Links ]
16. Pillebout E, Thervet E, Hill G, Alberti C, Vanhille P, Nochy D. Henoch-Schönlein Purpura in Adults: Outcome and Prognostic Factors. J Am Soc Nephrol 2002; 13:1271-1278. [ Links ]
17. Soyer T, Egritas O, Atmaca E, Akman H, Öztürk H, Tezic T. Acute pancreatitis: A rare presenting feature of Henoch Schonlein purpura. J Paediatr Child Health 2008; 44:152-153. [ Links ]
18. Fox M C, Carter S, Khouri I F, Giralt S A, Prieto V G, Nash J W, Hymes S R. Adult Henoch-Schönlein Purpura in a Patient With Myelodysplastic Syndrome and a History of Follicular Lymphoma. Cutis 2008; 81:131-137. [ Links ]
19. Fain O, Hamidou M, Cacoub P, Godeau B, Wechsler B, Paries J, et al. Vasculitides Associated With Malignancies: Analysis of Sixty Patients. Arthritis Rheum 2007; 57:1473-1480. [ Links ]
20. Blanco R M, González-Gay M A, Ibáñez D, López-Viana A, Ferrán C, Regueira A, González-Vela C. Henoch-Schönlein Purpura as Clinical Presentation of a Myelodysplastic Syndrome. Clin Rheumatol 1997; 16:626-628. [ Links ]
21. García-Porrua C, González-Gay M A. Cutaneous vasculitis as a paraneoplastic syndrome in adults. Arthritis Rheum 1998; 41:1133-1134. [ Links ]
22. Mitsui H, Shibagaki N, Kawamura T, Matsue H, Shimada S. A clinical study of Henoch-Schönlein Purpura associated with malignancy. JEAVD 2009; 23:394-401. [ Links ]
23. Okada J, Imafuku S, Tsujita J, Moroi Y, Urabe K, Furue M. Case of adult T-cell leukemia/lymphoma manifesting marked purpura. J Dermatol 2007; 34:782-785. [ Links ]
24. Blanco P, Denisi R, Rispal P, Deminière C, Pellegrin J L, Leng B, Aparicio M. Henoch-Schönlein purpura associated with segmental and focal proliferative glomerulonephritis in a patient with Hodgkin´s disease. Nephrol Dial Transplant 1999; 14:179-180. [ Links ]
25. Jean-Pierre Ng, J. Murphy, E. M. Chalmers, R. B. Hogg, R. L. Cumming, and S. Peebles. Henoch-Schönlein purpura and Hodgkin's disease. Postgrad Med J 1998; 64:881-882. [ Links ]
26. Day C, Savage C O S, Jones E L, Cockwell P. Henoch-Schönlein nephritis and non-Hodgkin´s lymphoma. Nephrol Dial Transplant 2001; 16:1080-1081. [ Links ]
27. Vesole D H. Diffuse large-cell lymphoma in an adult with Schönlein-Henoch purpura. Arch Intern Med 1987; 147:2026-2027. [ Links ]
28. Pertuiset E, Liote F, Launay-Russ E, Kemiche F, Cerf-Payrastre I, Chesneau A M. Adult Henoch-Schönlein Purpura Associated With Malignancy. Semin Arthritis Rheum 2000; 29:360-367. [ Links ]
29. Zurada J M, Ward K M, Grossman M E. Henoch-Schönlein purpura associated with malignancy in adults. J Am Acad Dermatol 2006; 55:S65-S70. [ Links ]

