










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Reumatología
Print version ISSN 0121-8123
Rev.Colomb.Reumatol. vol.20 no.2 Bogotá Apr./June 2013
Reporte de caso
Sarcoidosis en pediatría: reporte de 7 casos
Pediatric sarcoidosis: a report of seven cases
Ricardo Yépeza,*, Clara Malagóna y Carlos Olmosa
a Programa de Reumatología Pediátrica, Facultad de Medicina, Universidad El Bosque, Bogotá, Colombia
Recibido el 11 de febrero de 2012 Aceptado el 28 de marzo de 2013
* Autor para correspondencia. Correo electrónico: ricyepez@gmail.com (R. Yépez).
Resumen
La sarcoidosis es una enfermedad granulomatosa crónica de origen no infeccioso que puede comprometer diversos órganos. Su prevalencia es baja, y en Colombia se han reportado casos de manera aislada. Su etiología y fisiopatología aún no se conocen completamente. La presentación clínica y las diferentes manifestaciones de la enfermedad son variables. Cuando su debut se presenta en niños menores de 5 años se denomina sarcoidosis de inicio temprano, mientras que cuando lo hace en niños mayores de 5 años recibe el nombre de sarcoidosis de inicio tardío. En este reporte de caso se presentan 7 pacientes pediátricos, de los cuales 5 correspondieron a sarcoidosis de inicio temprano y 2 a sarcoidosis de inicio tardío. Todos los pacientes tuvieron un diagnóstico tardío de la enfermedad, manifestaciones de varios órganos y sistemas, y recibieron tratamiento inmunosupresor. Cuatro tuvieron curso crónico, 2 remisiones de la enfermedad y 1 recaídas frecuentes. Fue llamativa una asociación poco usual de 2 pacientes con sarcoidosis de inicio temprano quienes adicionalmente presentaron la enfermedad de Ollier.
Palabras clave: Sarcoidosis, Sarcoidosis en pediatría, Uveítis, Artritis, Enfermedad de Blau, Enfermedad de Ollier, Enfermedad granulomatosa, Sinovitis cauchosa.
Abstract
Sarcoidosis is a chronic granulomatous disease of non-infectious origin which can involve various target organs. Its prevalence is low and there have been only isolated cases reported in Colombia. Its etiology and pathophysiology are not well known. The clinical presentation and signs of the disease vary. When its onset is before five years of age it is recognized as early onset sarcoidosis, while if its onset is after five years of age it is called late onset sarcoidosis. In this case report all patients had a delayed diagnosis and they presented with a multiple organ involvement which required an immunosuppressive treatment. Of the 7 patients, 4 had a chronic course, 2 had remission, and 1 with frequent relapses of the disease. There was an unusual relationship of two patients with early onset disease who additionally presented with Ollier's disease.
Keywords: Sarcoidosis, Childhood sarcoidosis, Uveitis, Arthritis, Blau's disease, Ollier's disease, Granulomatous disease, Buggy synovitis.
Introducción
La sarcoidosis es una enfermedad granulomatosa crónica, con afectación de varios órganos y sistemas, y cuya etiología aún se desconoce1. A pesar de que afecta, principalmente, a adultos jóvenes, es poco frecuente en pediatría. Hoffman et al. (2004) describieron una incidencia de 0,29 por 100.000 niños/año, en menores de 15 años2. En Colombia, se han publicado varios reportes de caso de la enfermedad en población adulta pero solo uno incluyó población pediátrica3-9.
Frecuentemente, su diagnóstico se realiza en fases tardías de la enfermedad, en las cuales las manifestaciones sistémicas son más evidentes. En la población infantil se reconocen 2 formas distintas de sarcoidosis: sarcoidosis de inicio temprano (STEM) y sarcoidosis de inicio tardío (STAR). La STEM se caracteriza por presentarse en menores de 5 años y, usualmente, se manifiesta con la tríada de exantema cutáneo, uveítis y artritis, pero también se asocia a síntomas constitucionales y de otros órganos y sistemas. La STAR se expresa en niños mayores de 5 años, semejando la presentación clínica de sarcoidosis en los pacientes adultos, en los que predominan las manifestaciones pulmonares y linfadenopatías. La STAR se ha descrito con mayor frecuencia en la población afroamericana10,11.
Es importante sospecharla y reconocerla de manera temprana y oportuna para realizar un tratamiento adecuado de la misma, y disminuir, especialmente, sus complicaciones oculares y musculoesqueléticas.
A través de una revisión retrospectiva de los registros clínicos, de centros de referencia para reumatología pediátrica, en la ciudad de Bogotá (Fundación Cardioinfantil y consultorio de consulta externa de los autores), entre 1990 y 2011, se encontraron 7 casos en pacientes menores de 18 años que presentaron una clínica compatible con sarcoidosis y una confirmación histológica de los granulomas no caseificantes en los tejidos examinados. El diagnóstico de la enfermedad en todos los casos fue tardío con un tiempo promedio de 3,7 (1,5-6) años desde el inicio de los síntomas. Todos los pacientes recibieron tratamiento inicial para otras enfermedades, tales como tuberculosis pulmonar y cutánea, histiocitosis de células de Langerhans, artritis juvenil con uveítis, cistinosis y sinovitis villonodular. Adicionalmente, 2 de estos pacientes presentaron una asociación poco usual con enfermedad de Ollier. En este reporte de caso, 4 pacientes tuvieron un curso crónico, 2 remisiones de la enfermedad y 1 recaídas frecuentes de la enfermedad.
Presentación de los casos
El resumen de las principales características clínicas de este reporte de caso se presenta en las tablas 1 y 2.
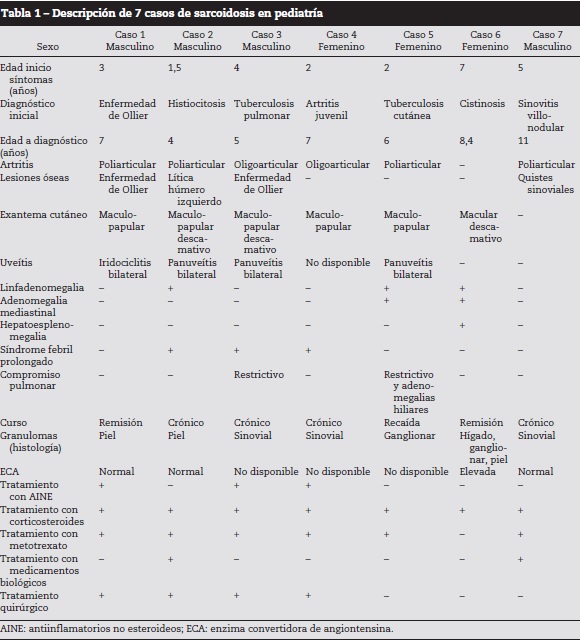
Caso 1
Paciente masculino quien desde los 3 años presentó artralgias en rodillas, piernas y tobillos, asociadas a lesiones maculopapulares no pruriginosas en tórax, abdomen y extremidades, e iridociclitis bilateral con pérdida progresiva de la agudeza visual. Se documentaron múltiples lesiones quísticas de falanges y húmero con una biopsia ósea compatible con enfermedad de Ollier (fig. 1). A los 7 años, se documentó artritis de rodillas, tobillos y muñecas, de consistencia cauchosa e indolora, con limitación leve a arcos de movimiento y agudización de lesiones cutáneas. Una biopsia de piel evidenció un proceso de inflamación crónica granulomatosa no caseificante. Se inició tratamiento inmunosupresor sistémico y tópico ocular con respuesta clínica satisfactoria.
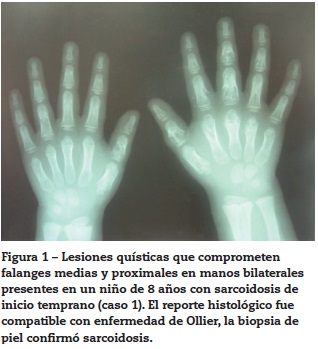
Caso 2
Paciente masculino quien a los 18 meses presentó cuadro febril prolongado asociado a lesiones hiperpigmentadas descamativas en pliegues cutáneos y pies, y también una lesión lítica en húmero izquierdo. Se realizó una biopsia de piel cuyo resultado fue un proceso granulomatoso que se interpretó y trató inicialmente como histiocitosis de células de Langerhans. A los 4 años de edad se evidenció poliartritis con presencia de pannus en ambas manos, reactivación de lesiones en piel y aparición de uveítis bilateral, por lo que se realizó una nueva biopsia en piel cuyo hallazgo fue una lesión granulomatosa compatible con sarcoidosis. Su curso fue el de un paciente corticodependiente y refractario a tratamiento con múltiples tratamientos inmunosupresores que incluyeron metotrexato, ciclosporina A, medicamentos biológicos (etanercept, infliximab, adalimumab) y que requirió múltiples intervenciones quirúrgicas oculares y articulares. En el último seguimiento, a los 17 años de edad, y a pesar del tratamiento inmunosupresor, presentó deformidades articulares, osteoporosis, cataratas y desprendimiento de retina bilateral.
Caso 3
Paciente masculino quien desde los 4 años presentó oligoartritis de rodillas y tobillos para lo cual recibió tratamiento con antiinflamatorios no esteroideos y corticoides. Posteriormente, estuvo hospitalizado por síndrome febril y compromiso pulmonar y ante sospecha de tuberculosis pulmonar se inició manejo con isoniacida. A los 5 años de edad se confirmó iridociclitis, la cual fue refractaria al tratamiento y requirió múltiples cirugías para corrección de sinequias y cataratas bilaterales. Se documentó enfermedad de Ollier confirmada con biopsia en lesión quística de segunda falange de segundo dedo de la mano derecha. Se realizó artrocentesis de rodilla izquierda, obteniendo líquido inflamatorio estéril, y la biopsia sinovial sugirió sarcoidosis. En la radiografía de tórax, se documentaron infiltrados pulmonares intersticiales y signos de hipertensión pulmonar y progreso a un patrón pulmonar restrictivo. A pesar del tratamiento con metotrexato y corticoide, la enfermedad continuó activa, con síntomas articulares y pulmonares que motivaron el uso de metotrexato endovenoso y una nueva sinovectomía de rodilla.
Caso 4
Paciente femenino quien a los 2 años presentó cuadros febriles intermitentes asociados a artralgias y cojera. A los 4 años se documentó artritis deformante en manos y muñecas, por lo que se inició tratamiento para artritis juvenil con sales de oro; a los 5 años de edad requirió corrección quirúrgica de deformidad en botonera y sinovectomía de muñeca. Presentó varios episodios de exantema en piel, en tronco, muslos y glúteos, y tos seca matinal. A los 6 años se realizó biopsia sinovial que documentó inflamación crónica granulomatosa no caseificante. Se realizaron sinovectomías múltiples y corrección de deformidad en dedos en cuello de cisne. A los 7 años de edad inició tratamiento con metotrexato y antiinflamatorios no esteroideos con buena respuesta terapéutica. A los 9 años de edad presentó reactivación clínica articular y cutánea. Se realizó nueva biopsia en piel, la cual fue compatible con sarcoidosis y se inició deflazacort.
Caso 5
Paciente femenino quien a los 2 años presentó lesiones maculopapulares en extremidades y en cuero cabelludo. El resultado de una biopsia de piel fue sugestivo de tuberculosis cutánea por lo que recibió tratamiento con ciclo corto de isoniacida, rifampicina y etambutol, presentando mejoría parcial. Consultó a reumatología por artritis en tobillos (fig. 2) asociada a linfadenopatías y disnea de grandes esfuerzos. Se evidenció adenomegalias múltiples en varios grupos regionales, marcado pannus y livedo reticularis. La radiografía y tomografía axial computarizada de tórax mostraron un infiltrado intersticial sin adenomegalias mediastinales (fig. 3), la curva de flujo volumen fue compatible con patrón restrictivo pulmonar y el lavado broncoalveolar descartó tuberculosis pulmonar. Se evidenció panuveítis bilateral. Se realizó una biopsia ganglionar que reportó granuloma no caseificante con tinción de Ziehl-Neelsen negativa e inmunofluorescencia negativa.
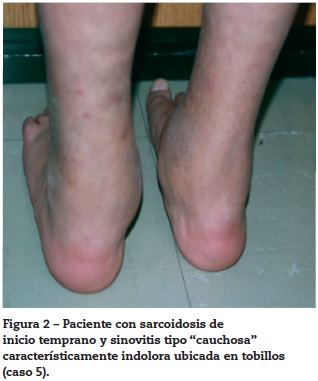
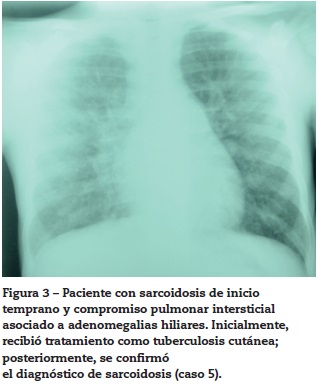
Se diagnosticó sarcoidosis y se inició manejo con corticoides y metotrexato, presentando respuesta favorable. La paciente tuvo mala adherencia al tratamiento con reactivación de la enfermedad.
Caso 6
Paciente femenino quien desde los 7 años presentó hepatoesplenomegalia, linfadenopatía ganglionar periférica, disnea y acrocianosis. Se asoció a baja talla y nefritis intersticial, por lo que se consideró una probable cistinosis. Posteriormente, se confirmó compromiso pulmonar intersticial y linfadenopatía parahiliar bilateral en tomografía de tórax, requiriendo tratamiento con oxígeno domiciliario. A los 8 años y 4 meses de edad aparecieron lesiones eritematosas multiformes de bordes bien definidos y patrón confluente, elevado y descamativo generalizado, de predominio en rostro, cuello, tronco y extremidades, que respetaba la zona genital y no era pruriginoso. Se asoció a xerodermia, opacidad corneal progresiva y persistencia de hepatoesplenomegalia y linfadenomegalia. Se realizaron biopsias de hígado, ganglio periférico y piel que evidenciaron proceso granulomatoso no caseificante y se descartó malignidad. Se confirmó compromiso hepatocelular no colestásico sin evidencia de hipertensión portal. La enzima convertidora de angiotensina (ECA) estuvo elevada. Se ordenó inicio de prednisolona con mejoría del compromiso cutáneo y disminución de la hepatoesplenomegalia. Durante 8 meses de seguimiento no hubo evidencia de compromiso ocular o articular.
Caso 7
Paciente masculino quien a los 5 años presentó poliartritis de rodillas, tobillos, muñecas y codos, con imagen de lesiones quísticas periarticulares compatibles con quistes sinoviales. Se realizó una biopsia sinovial que evidenció sinovitis crónica granulomatosa, la cual fue interpretada inicialmente como sinovitis villonodular. El compromiso poliartricular fue persistente, por lo que a los 11 años fue remitido a reumatología pediátrica. En el examen se evidenció poliartritis de consistencia cauchosa, indolora pero con arcos de movimiento conservados. Se descartó compromiso ocular, pulmonar y ganglionar. Los niveles de ECA fueron normales. Se inició manejo con corticoide oral y metotrexato. El paciente presentó mala adherencia al tratamiento médico y el curso de su enfermedad fue el de una enfermedad crónica activa a pesar del tratamiento.
Discusión
El International Registry of Sarcoid Arthritis in Childhood y el Pediatric Granulomatosus Arthritis, an International Registry son los registros de sarcoidosis pediátrica con el mayor número de pacientes en el mundo. Para el año 2000, el primero incluyó a 53 pacientes de 14 países diferentes, y para 2006, el segundo, a 61 pacientes12,13.
En Colombia, Valovis (1977) reporta 51 pacientes con sarcoidosis pulmonar en una revisión multicéntrica de 9 centros médicos en la ciudad de Bogotá, solo 10 pacientes estuvieron en el rango de edad entre los 10 y 20 años, y el autor realiza únicamente la revisión de 2 casos, ambos de sarcoidosis en adultos. De esta manera, este es uno de los reportes de caso de sarcoidosis en pediatría más grande publicado en Colombia y es el único que incluye pacientes con STEM3-9.
Este reporte presenta los casos que fueron referidos al servicio de reumatología pediátrica ante la sospecha de una enfermedad reumática, y consideramos que otros casos de sarcoidosis no son diagnosticados o estudiados de manera oportuna. A diferencia de lo descrito en la literatura, y a pesar de que el número de pacientes fue bajo, se observó una tendencia a mayor presentación de STEM (5/7) comparado con STAR (2/7), encontrando una distribución similar entre ambos sexos en los 2 grupos11,12.
Entre los pacientes con STEM, la tríada clásica de compromiso en piel, cutáneo y articular fue el hallazgo clínico predominante; 5/5 pacientes tuvieron exantema maculopapular, 3/5 poliartritis, 2/5 oligoartritis y 5/5 uveítis. Tres de cinco presentaron síndrome febril prolongado. El curso de la enfermedad fue crónico en 3/5, remisión de la enfermedad 1/5 y de recaídas en 1/5. El curso crónico que semejó artritis idiopática juvenil fue el más frecuente.
De los 2 casos con STAR, el primero desarrolló un compromiso del sistema reticuloendotelial sin evidencia de alteraciones oculares o articulares. El segundo paciente manifestó una poliartritis crónica con la característica sinovitis cauchosa (buggy synovitis) indolora y con escasa limitación de los arcos de movimiento de la sarcoidosis. Ninguno de los pacientes presentó la tríada de Löfgren (eritema nodosum, linfadenopatía bilateral y artritis), la cual es de frecuente presentación en sarcoidosis de la población adulta1,6,14. En STAR, el curso evidenciado fue el de remisión de la enfermedad y el de una enfermedad crónica en cada paciente, respectivamente.
A diferencia de lo reportado en la literatura, donde el compromiso pulmonar es más frecuente en STAR, en este reporte de caso los hallazgos de patrón pulmonar intersticial y restrictivo los manifestaron pacientes con STEM. Este hallazgo podría estar asociado al diagnóstico y tratamiento tardío de la enfermedad y es diferente a lo reportado por Shetty y Gedalia (2008) pero similar a lo descrito por Becker y Rose (2006)11,15.
Los hallazgos óseos de sarcoidosis que evidenciamos incluyeron lesiones líticas (caso 2) y quistes óseos (caso 7). Similar a lo descrito por Lee et al. (2012), se observó una asociación poco usual de 2 pacientes (casos 1 y 3) con STEM, quienes inicialmente presentaron lesiones quísticas múltiples, las cuales fueron biopsiadas confirmando un diagnóstico asociado de enfermedad de Ollier16. El hallazgo es llamativo y podría indicar la necesidad de realizar biopsia ósea en los pacientes con sarcoidosis que se manifiesten con lesiones óseas, con el fin de evaluar la posibilidad de enfermedad de Ollier asociada.
La enfermedad de Ollier es la encondroma to sis múltiple más frecuente y afecta predominantemente a niños en la primera década de la vida, con excepción del reporte de Lee no se ha asociado previamente a sarcoidosis16. Se manifiesta como masas óseas palpables de cartílago hialino maduro originadas en la médula de los huesos tubulares, especialmente de manos y pies. Las lesiones se caracterizan por su aparición múltiple, asimétrica, de predominio unilateral y sin un patrón hereditario conocido. El caso 1 presenta los hallazgos radiológicos característicos en falanges de dedos de las manos. Se requiere de un seguimiento clínico a largo plazo dado un riesgo de 35% de transformación sarcomatosa de las lesiones17.
Las claves para el diagnóstico de sarcoidosis son un alto índice de sospecha clínica, una búsqueda de los órganos blancos comprometidos y el análisis histológico de los granulomas no caseificantes típicos de la enfermedad. Otras enfermedades como la tuberculosis, granulomatosis de Wegener o silicosis pueden ser reconocidas y descartadas mediante el análisis histológico6. En este reporte de caso, la biopsia fue la herramienta más específica y útil; 3/7 en piel y 3/7 en tejido sinovial confirmaron el diagnóstico.
Una de las herramientas de laboratorio para el estudio de sarcoidosis es la medición de los niveles de ECA. Dado que su producción se origina en las células epitelioides del tejido granulomatoso sarcomatoso, sus niveles se relacionan con la carga total de granuloma corporal. Lindslay et al. (2000) encontraron una positividad de ECA en 38% de los pacientes valorados, mientras en este reporte de caso solo se encontró en 1 de 4 pacientes a los que se les realizó su medición1,12.
El diagnóstico diferencial de sarcoidosis debe incluir enfermedades infecciosas (tuberculosis e infecciones micóticas), enfermedades autoinmunes (artritis idiopática juvenil, lupus eritematoso sistémico, granulomatosis de Wegener, enfermedad de Crohn), otras enfermedades que se manifiestan por compromiso de varios órganos y sistemas y enfermedades neoplásicas (leucemia, linfoma). El síndrome de Blau debe incluirse en el diagnóstico diferencial de STEM1,12,13,15.
El síndrome de Blau fue descrito en 1985 en 11 miembros de un grupo familiar en el cual se evidenció un patrón de herencia autosómico dominante, asociado a una mutación en el gen NOD2 (antiguamente conocido como CARD15, ubicado en locus 16p12-21), característica que permite diferenciar esta entidad de STEM. El síndrome de Blau expresa un elemento de anticipación o empeoramiento de los síntomas en generaciones sucedáneas. Sus manifestaciones clínicas de una artritis granulomatosa asociada a uveítis y a manifestaciones cutáneas son las mismas que las de STEM; algunos autores las han considerado parte de una misma enfermedad, siendo STEM la variedad esporádica y el síndrome de Blau la variedad heredada15.
Debe considerarse la sarcoidosis en el estudio de los pacientes con uveítis autoinmune, masa mediastinal, enfermedad pulmonar intersticial, dermatitis crónica asociada con las manifestaciones de otros órganos y fiebre de origen desconocido. La alta prevalencia de uveítis (4/7) sugiere que la sarcoidosis debe ser incluida en su diagnóstico diferencial, especialmente en casos de panuveítis12.
Similar a los reportes en población pediátrica, y a diferencia del comportamiento de sarcoidosis en los adultos, en este reporte de caso, todos los pacientes requirieron tratamiento prolongado con corticoides y metotrexato, y su pronóstico fue pobre6,15. Mientras Milman et al. (2008) reportan un buen pronóstico de la enfermedad en población adulta18, en este reporte de caso, los tipos de curso clínico observados fueron el crónico, de remisión de la enfermedad y de recaídas y remisiones con enfermedad activa por muchos años. Solo 2 pacientes alcanzaron control sostenido de la enfermedad con el tratamiento. Similar a lo reportado en la literatura, la uveítis y la artritis se caracterizaron por un curso crónico: tres de cuatro pacientes con uveítis presentaron pérdida visual severa y requirieron procedimientos quirúrgicos oftalmológicos, 3/6 expresaron artritis crónica, 2/6 pacientes requirieron sinovectomía y 1/6 pacientes con artritis presentó una variedad deformante12,15,19.
Conclusiones
La sarcoidosis es una enfermedad poco frecuente en pediatría de origen granulomatoso crónico y de afectación de múltiples órganos y sistemas. Su incidencia, prevalencia y etiología aún no se conocen completamente. En el país existen solamente reportes de caso de la enfermedad.
La presencia de la tríada clásica de artritis, uveítis y manifestaciones cutáneas debe hacer pensar en STEM, mientras en este reporte de caso, los pacientes con STAR manifestaron un predominio de las manifestaciones de los sistemas reticuloendotelial y articular.
El compromiso pulmonar no es exclusivo de STAR y también puede presentarse en STEM, probablemente se asocie al diagnóstico y tratamiento tardíos de la enfermedad.
La biopsia del órgano blanco comprometido permitió la confirmación histológica y ayudó a descartar otros procesos granulomatosos crónicos, en especial la tuberculosis. El sitio preferido para la biopsia fue la piel seguida de la biopsia si novial.
Todos los pacientes requirieron tratamiento inmunosupresor y de largos períodos de seguimiento clínico.
El curso crónico con uveítis y poliartritis determinó un mal pronóstico.
Conflicto de intereses
Los autores declaran que no tienen ningún conflicto de intereses.
Agradecimientos
A Erika Lugo y Dolly García.
Bibliografía
1. Müller-Quernheim J, Prasse A, Zissel G. Pathogenesis of sarcoi dosis. Presse Med. 2012;41:e275-87. [ Links ]
2. Hoffmann AL, Milman N, Byg KE. Childhood sarcoidosis in Denmark 1979-1994: incidence, clinical features and laboratory results at presentation in 48 children. Acta Paediatr. 2004;93:30-6. [ Links ]
3. Velásquez CJ, Ramírez LA. Sarcoidosis cardíaca con compromiso pulmonar estadio IV. Rev Colomb Reumatol. 2006;13: 170-4. [ Links ]
4. Saavedra PG, Ramírez I, Cardona A. Hipercalcemia recurrente en un paciente con sarcoidosis aguda. Rev Colomb Reumatol. 2007;14:310-9. [ Links ]
5. Gómez-Puerta JA, Musuruana J, Sáez C, Cervera R, Font J. Sarcoidosis como espondiloartropatía seronegativa. Biomédica. 2005;25:435-8. [ Links ]
6. Valovis R. Sarcoidosis estudio clínico de 51 casos y revisión de la literatura. Acta Médica Colombiana. 1977;2:101-10. [ Links ]
7. Rocha OG, García PK, Enrique J, D'Achiardi RE, Rodríguez MP, Córdoba JP, et al. Sarcoidosis y compromiso renal: reporte de un caso y revisión de la literatura científica. Univ Méd Bogotá. 2012;53:94-102. [ Links ]
8. Polanía E, Morales G, Giraldo I, Caballero A. Sarcoidosis: manifestaciones radiológicas no usuales: presentación de un caso. Rev Colomb Neumol. 1990;1:77-80. [ Links ]
9. Izquierdo W, Rueda J. Sarcoidosis: cuatro presentaciones diferentes. Rev Colomb Reumatol. 1999;6:326-30. [ Links ]
10. Lazarus A. Sarcoidosis: epidemiology, etiology, pathogenesis, and genetics. Dis Mon. 2009;55:649-60. [ Links ]
11. Shetty AK, Gedalia A. Childhood sarcoidosis: A rare but fascinating disorder. Pediatr Rheumatol Online J. 2008;23:16. [ Links ]
12. Lindsley CB, Petty RE. Overview and report on international registry of sarcoid arthritis in childhood. Curr Rheumatol Rep. 2000;2:343-8. [ Links ]
13. Rosé CD, Wouters CH, Meiorin S, Doyle TM, Davey MP, Rosenbaum JT, et al. Pediatric granulomatous arthritis: an international registry. Arthritis Rheum. 2006;54:3337-44. [ Links ]
14. Suzuki E, Kanno T, Ohira H. Acute-onset sarcoidosis with polyarthralgia and hilar lymphadenopathy. Mod Rheumatol. 2010;20:188-92. [ Links ]
15. Becker ML, Rose CD. Blau syndrome and related genetic disorders causing childhood arthritis. Curr Rheumatol Rep. 2005;7:427-33. [ Links ]
16. Lee JH, Lim YJ, Lee S, Joo KB, Choi YY, Park CK, et al. Early-onset Childhood Sarcoidosis with Incidental Multiple Enchon dromatosis. J Korean Med Sci. 2012;27:96-100. [ Links ]
17. Silve C, Jüppner H. Ollier disease. Orphanet J Rare Dis. 2006;22:37. [ Links ]
18. Milman N, Hoffmann AL. Childhood sarcoidosis: long-term follow-up. Eur Resp J. 2008;31:592-8. [ Links ]
19. Blank N, Max R, Autschbach F, Libicher M, Lorenz H-M. Familial early onset sarcoidosis with bone cysts and erosions. Skeletal Radiol. 2007;36:891-3. [ Links ]
