










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Colombiana de Reumatología
versión impresa ISSN 0121-8123
Rev.Colomb.Reumatol. vol.22 no.3 Bogotá jul./sep./ 2015
https://doi.org/10.1016/j.rcreu.2015.05.003
http://dx.doi.org/10.1016/j.rcreu.2015.05.003
Informe de caso
Lupus eritematoso sistémico versus urticaria vasculítica hipocomplementémica: un dilema diagnóstico en la práctica clínica
Systemic lupus erythematosus versus hypocomplementemic urticarial vasculitis: a diagnostic dilemma in clinical practice
Viviana Parraa,*, Hernán Darío Aguirrea, Rodrigo Antonio Dazaa, Sergio Alexander Morab y Elkin Peñarandac
a Universidad de La Sabana, Hospital Universitario La Samaritana, Bogotá, Colombia
b Departamento de Reumatología, Hospital Universitario de la Samaritana, Bogotá, Colombia
c Departamento de Dermatología, Hospital Universitario de la Samaritana, Bogotá, Colombia
* Autor para correspondencia. Correo electrónico: vivipaz16@hotmail.com (V. Parra).
Historia del artículo: Recibido el 6 de febrero de 2015 Aceptado el 12 de mayo de 2015 On-line el 29 de junio de 2015
Resumen
El síndrome urticarial vasculítico hipocomplementémico forma parte de la vasculitis urticarial hipocomplementémica, presentándose en paciente con lesiones urticariales y compromiso sistémico e inmunológico, puede manifestarse de manera simultánea con una enfermedad del tejido conectivo como el lupus eritematoso sistémico o como una entidad independiente. Presentamos un caso de un paciente con características clasificatorias de lupus eritematoso sistémico, sin embargo, con presentación clínica de un síndrome urticarial vasculítico hipocomplementémico, realizando una revisión de la literatura, de sus posibles diferencias y similitudes.
Palabras clave: Vasculitis, Urticaria, Lupus eritematoso sistémico, Anti C1q.
Abstract
Hypocomplementemic urticarial vasculitis syndrome is part of the hypocomplementemic urticarial vasculitis, occurring in patients with urticarial lesions and systemic and immune compromise, which can appear simultaneously with a connective tissue disease such as systemic lupus erythematosus or an independent disorder. The case is presented of a patient with systemic lupus erythematosus qualifying features, however, with a clinical presentation of an hypocomplementemic urticarial vasculitis syndrome. A literature review was performed in order to present the potential differences and similarities.
Keywords: Vasculitis, Urticaria, Systemic lupus erythematosus, C1q Complement.
Introducción
El síndrome urticarial vasculítico hipocomplementémico (SUVH) hace parte de las vasculitis urticariales hipocomple-mentémicas, siendo una entidad con una presentación más severa, descrito por primera vez por McDuffie et al., en la Clínica Mayo en 19731,2. Caracterizado por lesiones urticariales con una presentación de más de 24 horas, asociado a compromiso sistémico renal, pulmonar, ocular, cardiaco y con alteraciones inmunológicas dadas por presencia de anticuerpos anti-Clq y disminución de los niveles de C1q por activación de la vía clásica del complemento3.
Es debatido el planteamiento de que el SUVH forma parte de las enfermedades del tejido conectivo, en cambio se plantea que puede ser una entidad independiente con riesgo a largo plazo de presentar una de estas enfermedades o asociarse de manera simultánea con ellas, sobre todo con el lupus eritematoso sistémico (LES)4, dado que comparten múltiples características y algunos autores, incluso, lo consideran un tipo inusual de LES. Se ha descrito que el SUVH está presente en 7 a 8% de los pacientes con LES y que el 54% de los pacientes con SUVH cumplen con criterios de LES al seguirlos en el tiempo5. Existen varios reportes de casos en la literatura de pacientes que cumplen tanto criterios de clasificación de LES como de SUVH6,7.
Teniendo en cuenta esta discusión realizamos una revisión de tema y planteamos un caso de un paciente con características clínicas e inmunológicas tanto de SUVH como de LES, considerando una fisiopatología que puede ser similar y sugiriendo que ambas entidades podrían formar parte de un mismo espectro de enfermedad autoinmune.
Caso clínico
Paciente masculino de 55 años, con antecedentes de enfermedad coronaria, neumopatía crónica en tratamiento con inhaloterapia y dosis de prednisona de 25 mg/día, por múltiples exacerbaciones, quien relata cuadro de 3 años de evolución, artralgias en pequeñas articulaciones, asociadas a lesiones tipo habón en tronco y extremidades, de duración de más de 24 horas no pruriginosas (figs. 1 y 2).
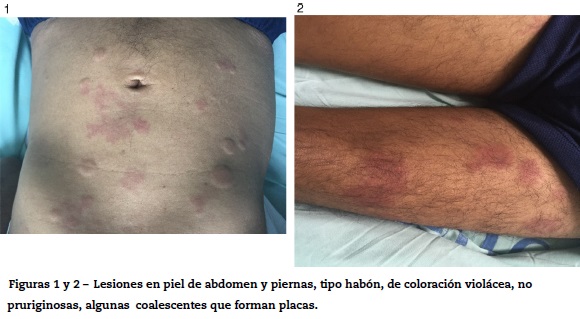
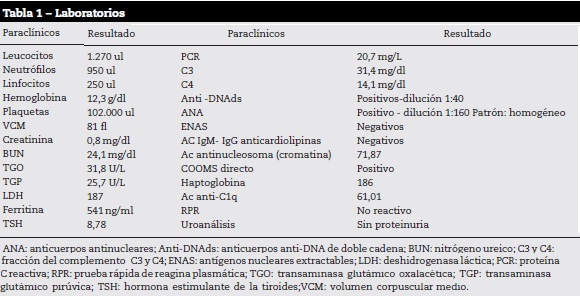
Narra que durante el primer año se le realizó biopsia de piel, que según él mostró un diagnóstico inicial de dermatosis neutrofílica, se solicitan paraclínicos (tabla 1), en los que llama la atención la presencia de bicitopenia, consumo de complemento, ANA positivos con patrón homogéneo, anti-DNAds positivo, Ac anticromatina y Ac anti-C1q positivos, ante la sospecha de neoplasia hematológica se realiza aspirado y biopsia de médula ósea (BMO) que no evidencia alteraciones de tipo neoplasia.
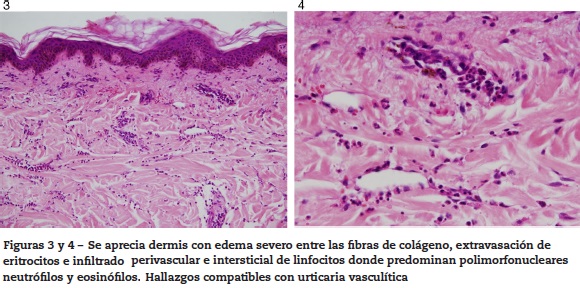
Ante la falta de claridad de diagnóstico previo, la presencia de nuevas lesiones en piel, la asociación con hipocomplementemia y presencia de marcadores de autoinmunidad, se decide realizar biopsia de las lesiones en piel (figs. 3 y 4) la cual describe hallazgos compatibles con urticaria vasculítica, configurando un cuadro clínico de urticaria vasculítica hipocomplementémica asociada a la presencia de ANA y anti-DNAds positivo, sin órgano mayor afectado, sin úlceras orales, ni alopecia, sin fotosensibilidad, sin fenómeno de Ray-naud, sin hematuria, sedimento urinario seriado normal, sin lesiones dérmicas típicas de lupus eritematoso, dejando en duda si las lesiones en piel son una entidad independiente, configurando un SUVH o hacen parte del complejo de manifestaciones atípicas o no específicas de lupus.
Discusión
El SUVH es una entidad rara dentro del tipo de vasculitis urticariales (VU), caracterizado por presentar lesiones urticariales no pruriginosas de más de 24 horas de evolución8, disminución de las fracciones C3, C4, CH50, C1q del complemento y presencia de anticuerpos contra la porción del C1q3,5,9 y manifestaciones sistémicas dadas por artralgia/artritis1,3,10, angioedema7,10, inflamación ocular con conjuntivitis, epiescleritis, uveitis7,8, glomerulonefritis3,10,11, EPOC3,12 y más raramente, pericarditis3.
En 1982 Schwartz estableció los criterios preliminares mayores y menores para hacer el diagnóstico de SUVH12 (tabla 2)6,13. El examen histológico muestra lesiones de tipo vasculitis leucocitoclástica, más a menudo con un predominantemente infiltrado neutrofilico pero sin interrupción de las paredes de los vasos9.

La incidencia de VU va del 2-20%14, sin embargo, es desconocida la incidencia del SUVH, en un estudio retrospectivo el 18% de los pacientes con VU presentaron biopsias hipocomplementémicas5. El SUVH afecta más a mujeres que a hombres, con una relación 2:1 y el pico de incidencia está alrededor de la quinta década de la vida, aunque también se han presentado reportes en niños9. Está presente en el 7 a 8% de los pacientes con LES, teniendo en cuenta que aproximadamente 250.000 estadounidenses tienen LES, esto indicaría que entre 17.500 y 20.000 americanos podrían tener el SUVH15.
La fisiopatología del SUVH no está clara, se han planteado múltiples mecanismos incluyendo la participación de depósitos de inmunocomplejos, activación del linfocito T y presencia de anticuerpos anti-C1q16. La presencia de inmunocomplejos representa una activación de la respuesta inmune humoral la cual puede ser responsable de la urticaria, el angioedema y la EPOC en el SUVH, los anticuerpos tipo IgG se unen contra la porción Fc de regiones similares al colágeno de la molécula C1, formando complejos inmunes y activando la vía clásica del complemento, generando gran cantidad de C3a y C5a17 y otras anafilotoxinas y quimioquinas que contribuyen a aumentar la permeabilidad vascular, quimiotaxis de células inflamatorias y depósito de complejos inmunes alrededor de los vasos18,19.
Los autoanticuerpos IgG2 anti-C1q se han encontrado en el 100% de SUVH y 35% LES, sugiriendo que estos anticuerpos se ligan al mismo epítope en estas dos entidades20'21, aunque algunos reportes evidencian que estas IgG pueden tener diferentes especificidades.
El mecanismo exacto del compromiso pulmonar en SUVH no está totalmente claro9, sin embargo, se detectan anticuerpos anti-Clq en EPOC, planteando que las moléculas de C1q se ligan a proteínas del surfactante pulmonar produciendo capi-laritis pulmonar, lo que contribuye a la enfermedad pulmonar obstructiva22, adicionalmente, también se ha encontrado sobreexpresión de elastasas de neutrófilo las cuales pueden hidrolizar proteínas del complemento y componentes del tejido conectivo en el SUVH3,23.
En cuanto a la activación del linfocito T, el C1q se ha asociado en la activación de inhibidores de las células T, adicionalmente los anti-Clq interfieren en la depuración de células apoptóticas, influyendo en la expresión de autoinmunidad al igual que en LES. En cuanto a la patogénesis de los anti-Clq está relacionada con la activación de la vía clásica del complemento y, adicionalmente, se han encontrado otras funciones biológicas incluyendo un papel modulador sobre las funciones celulares dentro de la respuesta adaptativa24, esta molécula se ha encontrado en menor porcentaje en personas sanas y en mayor porcentaje en pacientes con LES (61%), artritis reumatoide (20%), esclerodermia (15%), síndrome de Sjôgren (15%) y enfermedad mixta del tejido conectivo (15%)25. No está claro si estos anticuerpos tienen especificidad de unión al C1q, pero la evidencia sugiere que en el LES los anti-C1q se unen a la estructura terciaria intacta del C1q, mientras que en el SUVH se une a epítopes reducidos y desnaturalizados del C1q26,27.
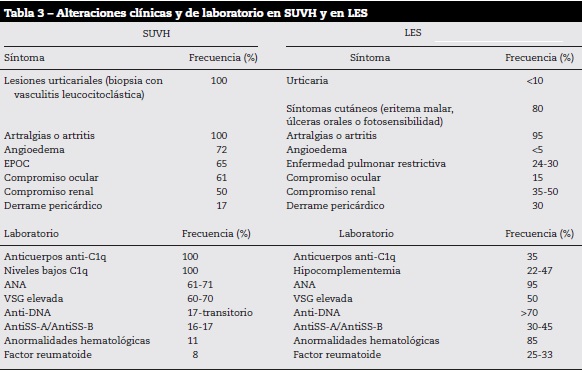
Dentro de las manifestaciones sistémicas, el SUVH y el LES pueden compartir características (tabla 3)3,4,6. La piel es el órgano dominante en el SUVH, con urticaria recurrente1,16, dolorosa, no pruriginosa, de más de 24 horas de evolución, con tendencia centrípeta, en tronco y extremidades que resuelven con hiperpigmentación postinflamatoria28. El angioedema puede presentarse hasta en el 50% de los pacientes comprometiendo labios, lengua, tejido periorbitario y manos, el cual es muy raro que se presente en el contexto de LES3,16. Este puede ser el primer signo del SUVH y sugiere compromiso de vasos profundos.
En la inmunohistoquímica se evidencian depósitos de Ig y complemento en las paredes de los vasos o en el endotelio, estos hallazgos se pueden presentar con vasculitis en LES, sin embargo, en LES presentan depósitos en la membrana basal con test de banda lúpica positivo9,14. Dentro del compromiso articular se pueden presentar artritis y artralgias en el 50% de los casos, el dolor es vago y transitorio y típicamente afecta codos, muñecas, rodillas y tobillos. La deformidad articular puede presentarse con artropatía de Jaccoud la cual se asocia a compromiso cardiaco con valvulopatía aórtica y mitral29,30.
El compromiso renal en SUVH es usualmente leve, se pueden encontrar proteinuria y hematuria14, en la histología puede presentar glomerulonefritis membranosa, membranoproliferativa o intra- o extracapilar, que no puede ser diferenciada del compromiso renal en LES16, por lo general el compromiso es más severo en niños, y cuando se presenta con falla renal terminal siempre debe buscarse otro diagnóstico diferencial. Un estudio con 18 pacientes con SUVH demostró 50% de compromiso renal, con manifestaciones que oscilaban desde mínima proteinuria a síndrome nefrótico con diferentes grados de hematuria y compromiso glomerular3.
El compromiso pulmonar puede ser muy variado, desde disnea, tos, hemoptisis, derrame pleural y EPOC, este último se ha asociado hasta en un 50% de los pacientes con SUVH3,28, presentándose 80-90% de anticuerpos anti-C1q3,20,31, el compromiso pulmonar, por lo general, se presenta en pacientes menores de 30 años y está relacionado con mayor morbi-mortalidad, el antecedente de tabaquismo puede empeorar el pronóstico, sin embargo, se puede presentar EPOC sin antecedente de uso de cigarrillo12,32.
El compromiso gastrointestinal es de alrededor del 30%, dado por dolor, náuseas, vómito, diarrea, ascitis asociado a serositis, hepatomegalia y esplenomegalia14,33. El 30% de los pacientes pueden presentar compromiso inflamatorio ocular, principalmente uveal, también hay reportes de conjuntivitis y epiescleritis1,3,9. El compromiso cardiaco, como ya se mencionó anteriormente, se ha encontrado en presencia de deformidad articular con compromiso valvular14,30.
Dentro de las características serológicas inmunológicas todos los pacientes con SUVH tienen anticuerpos anti-C1q asociados a lesiones de VU, pero raramente los pacientes con LES seropositivos para anti-C1q desarrollan lesiones de VU y, por el contrario, en el contexto del LES estos anticuerpos se correlacionan más con glomerulonefritis. A diferencia del LES en el cual casi el 100% de los pacientes tienen ANA positivos, solo entre el 50 y 60% de los pacientes con SUVH tienen ANA positivos en títulos moderados y en este último grupo de pacientes los anticuerpos anti-DNAds son transitorios e infrecuentes13.
En cuanto al tratamiento en los pacientes con SUVH, no hay ninguno específico, este se direcciona de acuerdo con el órgano y la severidad del cuadro. Los antihistamínicos típicamente se usan en urticaria, sin embargo son insuficientes para el manejo en SUVH, pueden utilizarse antiinflamatorios no esteroideos cuando hay compromiso articular6, sin embargo, se pueden utilizar diferentes tipos de inmunosupresores, similar al tratamiento del LES, como los corticosteroides, hidroxicloroquina, dapsona, e incluso ciclofosfamida, rituximab o inmunoglobulina, cuando hay un compromiso severo de la función renal o en casos refractarios3,7,9,12,20,34,35.
Conclusión
Con el planteamiento de este caso, se evidencia la dificultad en la diferenciación del SUVH con algunos tipos de presentación del LES, sin embargo, el reto diagnóstico no debe ser clasificar al paciente entre un síndrome o el otro, sino entender las similitudes fisiopatológicas y clínicas de estas dos entidades y de esta manera realizar un abordaje terapéutico temprano y adecuado, que permita un seguimiento clínico para determinar si estamos ante una entidad independiente o un síndrome de poliautoinmunidad.
Responsabilidades éticas
Protección de personas y animales. Los autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datos. Los autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informado. Los autores declaran que en este artículo no aparecen datos de pacientes.
Financiación
No se recibió financiación.
Conflicto de intereses
Los autores declaran no tener ningún conflicto de intereses.
Bibliografía
1. McDuffie FC, Sams WM Jr, Maldonado JE, Andreini PH, Conn DL, Samayoa EA. Hypocomplementemia with cutaneous vasculitis and arthritis Possible immune complex syndrome. Mayo Clin Proc. 1973;48(5):340-8. [ Links ]
2. Maldonado JE, Iglesias-Gamarra A. Vasculitis urticarial hipocomplementémica: aclaración histórica. Revista Colombiana de Reumatologia. 2014;21(2):84-90. [ Links ]
3. Wisnieski JJ, Baer AN, Christensen J, Cupps TR, Flagg DN, Jones JV, et al. Hypocomplementemic urticarial vasculitis syndrome Clinical and serologic findings in 18 patients. Medicine (Baltimore). 1995;74(1):24-41. [ Links ]
4. Trendelenburg M, Courvoisier S, Spath PJ, Moll S, Mihatsch M, Itin P, et al. Hypocomplementemic urticarial vasculitis or systemic lupus erythematosus. Am J Kidney Dis. 1999;34(4):745-51. [ Links ]
5. Davis MD, Daoud MS, Kirby B, Gibson LE, Rogers RS III. Clinicopathologic correlation of hypocomplementemic and normocomplementemic urticarial vasculitis. J Am Acad Dermatol. 1998;38 6 Pt 1:899-905. [ Links ]
6. Aydogan K, Karadogan SK, Adim SB, Tunali S. Hypocomplementemic urticarial vasculitis: a rare presentation of systemic lupus erythematosus Int J Dermatol. 2006;45(9):1057-61. [ Links ]
7. Saigal K, Valencia IC, Cohen J, Kerdel FA. Hypocomplementemic urticarial vasculitis with angioedema, a rare presentation of systemic lupus erythematosus: rapid response to rituximab. J Am Acad Dermatol. 2003;49 5 Suppl:S283-5. [ Links ]
8. Maurer M, Grabbe J. Urticaria: its history-based diagnosis and etiologically oriented treatment. Dtsch Arztebl Int. 2008;105(25):458-65, quiz 65-6. [ Links ]
9. Mehregan DR, Hall MJ, Gibson LE. Urticarial vasculitis: a histopathologic and clinical review of 72 cases. J Am Acad Dermatol. 1992;26 3 Pt 2:441-8. [ Links ]
10. Renard M, Wouters C, Proesmans W. Rapidly progressive glomerulonephritis in a boy with hypocomplementaemic urticarial vasculitis. Eur J Pediatr. 1998;157(3):243-5. [ Links ]
11. Ramirez G, Saba SR, Espinoza L. Hypocomplementemic vasculitis and renal involvement. Nephron. 1987;45(2):147-50. [ Links ]
12. Schwartz HR, McDuffie FC, Black LF, Schroeter AL, Conn DL. Hypocomplementemic urticarial vasculitis: association with chronic obstructive pulmonary disease. Mayo Clin Proc. 1982;57(4):231-8. [ Links ]
13. Rivas González AM, Universidad Pontificia Bolivariana M, Colombia, Velásquez Franco CJ, Universidad Pontificia Bolivariana M, Colombia, Pinto Peñaranda LF, Universidad Pontificia Bolivariana M, Colombia, et al. Urticarial Vasculitis. RevColombReumatol. 2009; 16(2):154-66. [ Links ]
14. Grotz W, Baba HA, Becker JU, Baumgartel MW. Hypocomplementemic urticarial vasculitis syndrome: an interdisciplinary challenge. Dtsch Arztebl Int. 2009;106(46):756-63. [ Links ]
15. Helmick CG, Felso DT, Lawrence RC, Gabriel S, Hirsch R, Kwoh CK, et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part I Arthritis Rheum. 2008;58(1):15-25. [ Links ]
16. Jara LJ, Navarro C, Medina G, Vera-Lastra O, Saavedra MA. Hypocomplementemic urticarial vasculitis syndrome. Curr Rheumatol Rep. 2009;11(6):410-5. [ Links ]
17. Chew GY, Gatenby PA. Inflammatory myositis complicating hypocomplementemic urticarial vasculitis despite on-going immunosuppression. Clin Rheumatol. 2007;26(8):1370-2. [ Links ]
18. Marder RJ, Burch FX, Schmid FR, Zeiss CR, Gewurz H. Low molecular weight C1q-precipitins in hypocomplementemic vasculitis-urticaria syndrome: partial purification and characterization as immunoglobulin. J Immunol. 1978;121(2):613-8. [ Links ]
19. Marder RJ, Potempa LA, Jones JV, Toriumi D, Schmid FR, Gewurz H. Assay, purification and further characterization of 7S C1q-precipitins (C1q-p) in hypocomplementemic vasculitis urticaria syndrome and systemic lupus erythematosus. Acta Pathol Microbiol Immunol Scand Suppl. 1984;284:25-34. [ Links ]
20. Wisnieski JJ, Jones SM. Comparison of autoantibodies to the collagen-like region of C1q in hypocomplementemic urticarial vasculitis syndrome and systemic lupus erythematosus. J Immunol. 1992;148(5):1396-403. [ Links ]
21. Wen L, Atkinson JP, Giclas PC. Clinical and laboratory evaluation of complement deficiency. J Allergy Clin Immunol. 2004;113(4):585-93, quiz 94. [ Links ]
22. Friskel E, Foster R. A 37-year-old man with severe COPD, rash, and conjunctivitis. Chest. 2000;118(5):1493-5. [ Links ]
23. Jamison SC, Brierre S, Sweet J, de Boisblanc B. A case of precocious emphysema and lung cancer in a woman with a history of hypocomplementemic urticarial vasculitis. Chest. 2008;133(3):787-9. [ Links ]
24. Sontheimer RD, Racila E, Racila DM. C1q: its functions within the innate and adaptive immune responses and its role in lupus autoimmunity. J Invest Dermatol. 2005;125(1):14-23. [ Links ]
25. Lienesch DW, Sherman KE, Metzger A, Shen GQ. Anti-Clq antibodies in patients with chronic hepatitis C infection. Clin Exp Rheumatol. 2006;24(2):183-5. [ Links ]
26. Antes U, Heinz HP, Loos M. Evidence for the presence of autoantibodies to the collagen-like portion of C1q in systemic lupus erythematosus. Arthritis Rheum. 1988;31(4):457-64. [ Links ]
27. Uwatoko S, Mannik M. Low-molecular weight C1q-binding immunoglobulin G in patients with systemic lupus erythematosus consists of autoantibodies to the collagen-like region of C1q. J Clin Invest. 1988;82(3):816-24. [ Links ]
28. Davis MD, Brewer JD. Urticarial vasculitis and hypocomplementemic urticarial vasculitis syndrome. Immunol Allergy Clin North Am. 2004;24(2):183-213, vi. [ Links ]
29. Sturgess AS, Littlejohn GO. Jaccoud's arthritis and panvasculitis in the hypocomplementemic urticarial vasculitis syndrome. J Rheumatol. 1988;15(5):858-61. [ Links ]
30. Palazzo E, Bourgeois P, Meyer O, de Bandt M, Kazatchkine M, Kahn MF. Hypocomplementemic urticarial vasculitis syndrome, Jaccoud's syndrome, valvulopathy: a new syndromic combination. J Rheumatol. 1993;20(7):1236-40. [ Links ]
31. Lee P, Gildea TR, Stoller JK. Emphysema in nonsmokers: alpha 1-antitrypsin deficiency and other causes. Cleve Clin J Med. 2002;69(12):928-9, 33, 36 passim. [ Links ]
32. Janoff A. Elastase in tissue injury. Annu Rev Med. 1985;36:207-16. [ Links ]
33. Sánchez NP, Winkelmann RK, Schroeter AL, Dicken CH. The clinical and histopathologic spectrums of urticarial vasculitis: study of forty cases. J Am Acad Dermatol. 1982;7(5):599-605. [ Links ]
34. Balsam L, Karim M, Miller F, Rubinstein S. Crescentic glomerulonephritis associated with hypocomplementemic urticarial vasculitis syndrome. Am J Kidney Dis. 2008;52(6):1168-73. [ Links ]
35. Fortson JS, Zone JJ, Hammond ME, Groggel GC. Hypocomplementemic urticarial vasculitis syndrome responsive to dapsone. J Am Acad Dermatol. 1986;15 5 Pt 2:1137-42. [ Links ]
