











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
Las vasculitis es un grupo de enfermedades que afectan la pared vascular mediante un proceso inflamatorio y son clasificadas tradicionalmente bajo el consenso de Chapel Hill, según el tipo de vaso sanguíneo que involucre en vasculitis de grandes vasos, vasos de mediano calibre y vasculitis de pequeños vasos. Esta última constituye una poliangeítis necrosante que afecta predominantemente a vénulas, arteriolas y capilares, sin embargo, puede involucrar arterias y puede presentarse sin que esté de por medio un mecanismo mediado por inmunocomplejos, denominadas por esta razón vasculitis pauciinmunes1.
Las vasculitis pauciinmunes, comúnmente están asociadas a la presencia de auto anticuerpos circulantes conocidos como anticuerpos anticitoplasma de neutrófilo (ANCA) dirigidos contra antígenos denominados proteinasa 3 o mieloperoxidasa que están presentes en los grânulos de los neutrófilos y monocitos. Este tipo de vasculitis constituyen formas clínicas localizadas teniendo como blanco renal al glomérulo, desarrollando una glomerulonefritis aguda, denominándose vasculitis limitada a riñón o pueden presentarse bajo una forma sistémica afectando múltiples órganos y tejidos, entre los que involucra al riñón, agrupándose bajo 3 subtipos bien definidos: granulomatosis de Wegener (GW), síndrome de Churg-Strauss y poliangeítis microscópica (PAM). El diagnóstico de estos diferentes subtipos de vasculitis pauciinmune de pequeños vasos, se facilita basándose en el síndrome acompañante2, así:
La GW ocurre en asociación a inflamación granulomatosa necrosante de cualquier parte del tracto respiratorio.
Síndrome de Churg-Strauss es una inflamación granulo-matosa necrosante que ocurre en asociación con asma y eosinofilia.
La PAM ocurre sin inflamación granulomatosa necrosante, sin asma ni eosinofilia.
Existe sintomatología constitucional que las acompaña, como fiebre, mialgias, malestar general, debilidad y anemia. Sin embargo, pueden ser completamente asintomáticas y sus manifestaciones ser secundarias al deterioro en la función renal en la forma localizada.
En México, no existe una publicación sobre serie de casos de vasculitis renal ANCA asociada, únicamente casos descritos aislados y estudios de vasculitis cutáneas, respiratorias; por tanto, nuestro estudio derivado de población mexicana, constituye un esfuerzo por mostrar el comportamiento epimanifestacionesdemiológico, clínico, con sus hallazgos histopatológicos y evolución de la vasculitis renal.
Materiales y métodos
Estudio descriptivo, retrospectivo, en el que se revisaron los casos atendidos en el área de nefrología de un hospital de tercer nivel durante 4 años, enero de 2008 a diciembre de 2012, desde su historial clínico recabado al momento del ingreso, su exploración física, hallazgos de laboratorio y gabinete, incluido su respectivo estudio inmunológico, biopsia renal, abordaje terapéutico y evolución clínica a 12 meses posdiagnóstico. Los criterios de inclusión fueron:
- Deterioro en la función renal, definido como un incremento del valor de creatinina >0,3 mg/dl de su valor basal conocido o un valor de creatinina sérica >1,3 mg/dl, con o sin oliguria.
Sedimento urinario activo.
Proteinuria en análisis de orina.
Hombres y mujeres de cualquier edad.
ANCA positivos.
Biopsia renal compatible con glomerulonefritis pauciinmune.
Se excluyeron pacientes conocidos con nefropatía previa, con lupus eritematoso sistémico, sin datos histológicos compatibles y ausencia de estudios inmunológicos. Se eliminaron pacientes que perdieron su seguimiento en el transcurso del primer año, irregulares en sus consultas y con poco apego al tratamiento prescrito.
Se realizó el análisis descriptivo con medidas de tendencia central y rangos para las variables continuas y cálculo de frecuencias para las variables categóricas.
Resultados
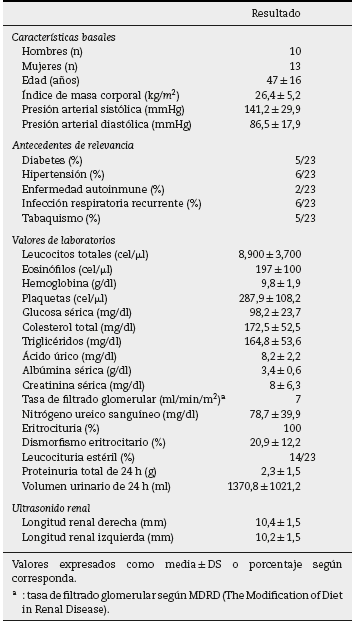
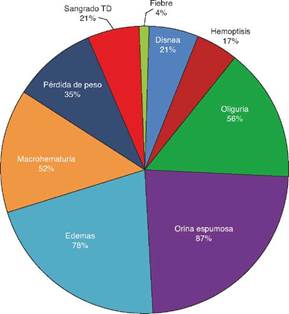
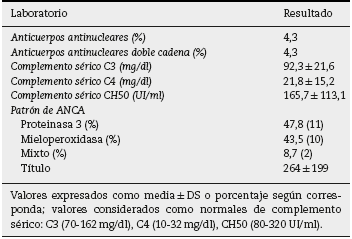
Basándonos en los criterios de inclusión propuestos, se seleccionaron 23 pacientes entre enero de 2008 y diciembre de 2012, 13 mujeres y 10 hombres, guardando una relación de 1,3:1, con una edad promedio de 47 ± 16 años, siendo el paciente de menor edad de 14 añosyde70años el mayor, con un índice de masa corporal promedio de 26,4 ± 5,2kg/m2, 21,7% de ellos con diabetes mellitus, 26,1% con hipertensión arterial sistémica y 8,7% con una enfermedad autoinmune subyacente controlada, tipo artritis reumatoide y cirrosis biliar primaria (tabla 1). El tiempo de evolución de su sintomatología renal al momento de su consulta fue en promedio de 1 a 3 meses para el 74% de los pacientes, 21,7% más de 3 meses; en su historial clínico se destaca la presencia de sintomatología constitucional en el 100% de los pacientes, 87% con orina espumosa, 78% con edemas, 56,5% con oliguria, macro-hematuria en 52%, la pérdida de peso referida en el 35% de los casos, teniendo alrededor del 20% datos de sangrado de tubo digestivo, disnea y hemoptisis (fig. 1). Los resultados de laboratorios generales reportaban una hemoglobina promedio de 9,8 ± 1,9 g/dl, un nivel de colesterol de 172,5 ± 52,5 mg/dl, triglicéridos de 164,8 ± 53,6 mg/dl, albúmina sérica de 3,4 ± 0,6 g/dl, una creatinina sérica de 8 ± 6,3 mg/dl, con una tasa de filtrado glomerular de 7 ml/min/m2, uremia de 78,7 ± 39,9 mg/dl; el análisis de orina con microhematuria en el 100% de los pacientes, pero con un dismorfismo eritrocitario promedio de 20,9 ± 12,2%, 61% con leucocituria estéril y un promedio de proteinuria de 24 h de 2,3 ± 1,5 g; la anatomía renal por ecografía reportaba longitudes renales en límites normales. El estudio inmunológico de los pacientes (tabla 2), incluyó la búsqueda de anticuerpos antinucleares (ANA) y anti-DNAds los que se encontraron presentes en uno de los pacientes, el complemento sérico C3 con un promedio de 92,3 ± 21,6 mg/dl, complemento sérico C4 con 21,8 ± 15,2 mg/dl, complemento sérico CH50 con 165,7 ± 113,1 UI/ml; se solicitaron anticuerpos ANCA a todos los pacientes, encontrando cuantitativamente un título de 264 ± 199, cuyas especificidades fueron para proteinasa 3 en un 47,8%, mieloperoxidasa en un 43,5% y con un patrón mixto el 8,7%.
En lo que respecta al estudio de biopsia renal (tabla 3), el promedio de glomérulos por muestra fue de 19, más del 50% de los glomérulos muestreados por biopsia tenía proliferación extracapilar, con una esclerosis global en el 30% y esclerosis segmentarias en el 26%, fibrosis intersticial de 34 ± 11%. El 100% de las biopsias tenía semilunas: celulares 2/23, fibrosas 2/23 y mixtas 19/23, arteriolopatía moderada a severa en 19/23, necrosis fibrinoide en 12/23, arteritis en 4/23 (figs.2 y 3).
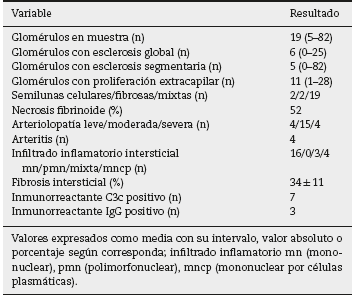
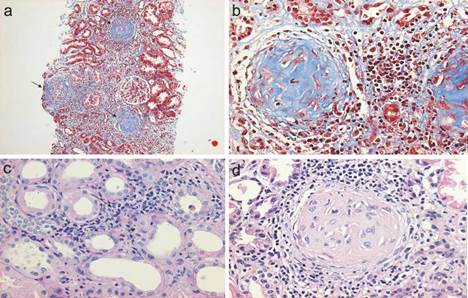
Figura 2 a) Biopsia renal en la que se muestra una vista panorámica (10X) de un cilindro de tejido con glomérulos esclerosados (tricrómico de Masson), en la que se pintan de color azul las fibras de colágeno que sustituyen al tejido glomerular normal b) con una extensa reacción inflamatoria periglomerular y peritubular intersticial mononuclear y polimorfonuclear que finalizan conformando una semiluna fibrocelular (c) y (d).
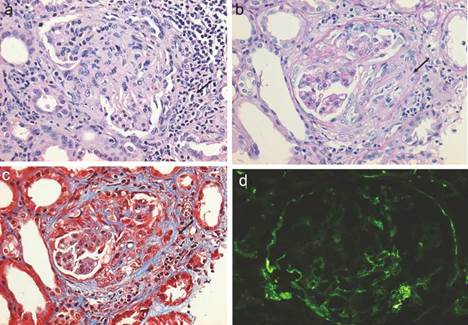
Figura 3 a) Glomérulo con severo infiltrado inflamatorio intersticial y periglomerular (tinción hematoxilina-eosina) que provoca trasudado de fibrina formando semilunas celulares que evolucionan a fibrocelulares b), las que al paso de pocas semanas terminan como semilunas fibrosas con extensa esclerosis glomerular acompañante c). Se muestra inmunofluorescencia de C3c con ausencia de un patrón granular fluorescente intenso como en las glomerulopatías autoinmunes, de ahí el término de pauciinmune (d).
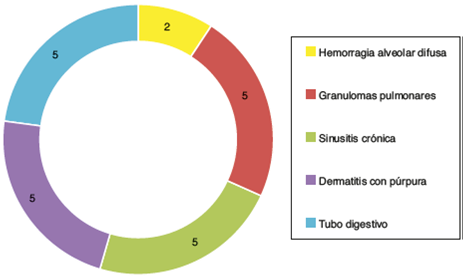
De las manifestaciones sistémicas 5/23 presentaron inflamación granulomatosa pulmonar, 2/23 con hemorragia alveolar difusa, 5/23 con sinusitis crónica granulomatosa, vasculitis cutánea en 5/23, así como hallazgos de vasculitis de tubo digestivo en 5/23 (fig. 4). Basados en los hallazgos clínicos, laboratorios, gabinete e histología, fueron catalogados como GW 10/23, poliangeítis microscópica 5/23, vasculitis limitada arinón 5/23 y síndrome Churg-Strauss 3/23. En la inmunofluorescencia se encontró positividad para inmunorreactante C3c en 7/23 e IgG en 3/23, sin embargo, es importante mencionar que no en el patrón característico granular por inmunocomplejos, sino como una tinción de baja intensidad.
Todos los pacientes recibieron una intervención terapéutica (tabla 4), siendo necesaria la terapia sustitutiva de la función renal en 15/23 pacientes debido al sínmanifestacionesdrome urémico y anuria; se usó terapia inmunosupresora basada en corticosteroides/ciclofosfamida en 16/23, corticos-teroides/ciclofosfamida/micofenolato de mofetilo en 4/23 y monoterapia con corticosteroides en 3/23, 5 pacientes fueron sometidos a plasmaféresis además de inmunosupresión basal. El desenlace final de los pacientes con vasculitis renal ANCA asociada a 12 meses, fue de 3 defunciones por complicaciones de la misma vasculitis, 15/23 en terapia dialítica crónica y 5/23 con terapia inmunosupresora libre de diálisis, con un estadio 4 de enfermedad renal crónica (TFG15-29 ml/min/m2).

Discusión
La causa de la vasculitis sistémica ANCA-asociada probablemente sea multifactorial, ya que se mantiene desconocida a la fecha, sin embargo, al paso de los años se han estudiado muchos factores asociados de tipo étnicos, geográficos, ambientales, exposición a fármacos y genéticos, a pesar de lo cual sin una intervención médica oportuna sigue teniendo una alta mortalidad a un año, cercana al 80% derivada de las múltiples complicaciones que ocasiona. Dicha mortalidad se ha logrado reducir con el uso de la terapia inmunosupresora que permite controlar la actividad de la enfermedad y limitar el daño a los órganos involucrados2,3.
Epidemiología de las vasculitis sistémicas
La vasculitis pauciinmune tiene una incidencia anual de alrededor de 5-20 casos por millón de habitantes y varía de una región a otra4. Las vasculitis sistémicas se presentan con mayor frecuencia a una edad adulta, de la quinta a la séptima década de la vida, sin embargo, las glomerulonefritis pauciinmunes con patrón rápidamente progresivo, son las formas más comunes de GN crescénticas a todas las edades, incluidas las personas de edad avanzada. En nuestra serie de casos, aunque el promedio de edad sobrepasaba los 40 años, tuvimos pacientes con edades extremas de 14 hasta 70 años, sin que estos tuvieran un antecedente como normalmente suele ocurrir, lo que demuestra que las vasculitis no discriminan edad. No existe un patrón de transferencia hereditaria que permita el desarrollo de la GW en más de un miembro de la familia, sin embargo, se han descrito asociaciones con algunos antígenos HLA DR2, HLA B7 y HLA DR1 sin que exista mayor explicación. La literatura describe una mayor prevalencia en población caucásica que en afroamericanos, así como una tendencia a mayores casos de GW en climas fríos y de PAM en climas cálidos5-7. De igual manera, con un mecanismo incierto desconocido, se describe el contacto crónico con sílice como un factor ambiental de riesgo para el desarrollo de vasculitis de pequeños vasos; también la exposición crónica a drogas como el propiltiouracilo, penicilamina y minociclina, se ha asociado a vasculitis con altos títulos de anticuerpos contra mieloperoxidasa (P-ANCA) (8-10. Al respecto, nuestra población de origen caucásico y proveniente de todo el país, aunque la mayoría del Valle de México con una altitud que supera los 2.200 msnm, donde las temperaturas son más frías, también, los familiares cercanos de nuestros pacientes fueron estudiados como parte de la investigación de cada caso, sin encontrar anormalidades que justificaran mayor intervención en los mismos.
Sitios de afección de vasculitis sistémicas
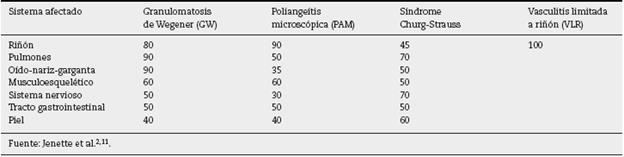
Aunque las primeras manifestaciones de las vasculitis sismanifestacionestémicas suelen ser un síndrome similar a influenza, con mialgias, artralgias, anorexia, pérdida de peso, fiebre, malestar general, etc., no constituyen la causa principal de sus consultas, hasta que un órgano específico se afecta11. Así, en nuestros pacientes el tiempo de consulta fue tardío en la mayoría de los casos, con un tiempo de evolución de sintomatología renal de uno a 3 meses, lo que marca el pronóstico por el nivel de daño glomerular encontrado en las biopsias, considerando sus características patogénicas de ser necrosante y rápidamente progresiva por destrucción tisular extensa. En la actualidad ya es conocido que las vasculitis de pequeños vasos pauciinmunes pueden afectar cualquier órgano, pero la frecuencia o proporción de los mismos es variable según el tipo de vasculitis (tabla 5) (2,12. De igual forma, existe una variedad de presentaciones reportadas en cada órgano o tejido afectado (tabla 6) (2, nosotros pudimos documentar al menos 3 sitios de vasculitis extrarrenal en los pacientes, algunos de ellos coexistiendo, por ejemplo: vasculitis renal con granulomas pulmonares, sinusales y en tubo digestivo, lo cual no guardaba correlación con el título de anticuerpos anticitoplasma ni con sus respectivas especificidades presentes

El compromiso renal en la vasculitis ANCA es frecuente, en general, la literatura la describe en el 50% de los pacientes en el momento del diagnóstico y entre el 70 y el 85% en el curso de la enfermedad13. El compromiso renal es muy común en GW y PAM, menos frecuente en síndrome de Churg-Strauss, siendo hematuria, proteinuria y falla renal las principales. Esta última con un patrón rápidamente progresivo y de características severas en GW y PAM, cerca de 40 a 95% de las GW tienen compromiso renal a su presentación y 64 a 100% en PAM3; en nuestra serie el 100% de la población tenía eritrocituria con un dismorfismo del 20%, lo que se considera bajo tomando en cuenta el tipo de glomerulonefritis en cuestión, sin embargo, el nivel de lesión renal era alto con una tasa de filtrado glomerular promedio de 7 ml/min/m2 y necesidad de terapia dialítica de urgencia en 65% de los mismos, lo cual fue confirmado con los hallazgos en sus biopsia renales con el alto grado de fibrosis y extensa necrosis de paredes vasculares glomerulares. A lo largo del tiempo, aunque recibieron terapia inmunosupresora todos los pacientes, la sobrevida renal fue baja, ya que únicamente el 21,7% (5 pacientes) permaneció libre de diálisis a 12 meses, manteniendo una tasa de filtrado glomerular entre 15 a 29 ml/min/m2, catalogándose dentro del estadio 4 de enfermedad renal crónica según la clasificación clásica propuesta por The National Kidney Foundation Kidney Disease Outcomes Quality Initiative, lo que finalmente es un pronóstico sombrío a corto plazo en su evolución clínica.
Opciones de tratamiento
El tratamiento debe guiarse por la premisa de «no sobretratar una enfermedad leve, ni infratratar una enfermedad severa»; la indicación de tratamiento inmunosupresor completo es con una glomerulonefritis severa que cause deterioro en la función renal, por lo que el tratamiento involucra 3 fases: inducción a la remisión, mantenimiento de la remisión y tratamiento de la recaída14.
Con la introducción de los agentes alquilantes como la ciclofosfamida, el pronóstico mejoró con el incremento en la sobrevida renal de 20-60% a un año con corticosteroides solos, hasta 87% a 8 años y 64% a 10 años, por lo que en la actualidad los regímenes más efectivos para inducción a la remisión incluyen ambas drogas(3,15,16. Una vez alcanzada la remisión, se usan fármacos con menos toxicidad para mantener la remisión, tales como azatioprina, metotrexato a bajas dosis, micofenolato de mofetilo y ciclosporina. Las tasas de recaídas son más bajas en la PAM que en la GW.
En nuestra población de estudio, todos fueron sometidos a la primera fase de inducción a la inmunosupresión, pero no todos fueron candidatos a continuar con la misma, sobre todo después de realizar la biopsia renal y reclasificar a cada uno de los pacientes según hallazgos histológicos, restando pronóstico de su función renal lo que obligó a reducir la inmunosupresión y posteriormente suspenderla. De esta forma, los pacientes recibieron un esquema diferente de inmunosupresión, definido con base en criterio clínico e histológico, algunos solamente corticosteroides (3/23), otros corticosteroides combinados con alquilantes tipo ciclofosfamida (16/23) y a otros se les adicionó además de lo anterior micofenolato de mofetilo como tercera droga (4/23), aquí es importante mencionar que se tuvo acceso a terapias como plasmaféresis y rituximab, las que fueron indicadas según la valoración del consenso de los especialistas en nefrología de la institución.
Factores de mal pronóstico en vasculitis renal ANCA
Los factores pronósticos aceptados son la velocidad con la cual se inicia el tratamiento, la función renal al momento del diagnóstico y la extensión del compromiso en la biopsia renal13. En la última década se han estudiado y definido otros factores adicionales que les confieren a las vasculitis ANCA un pronóstico sombrío, entre ellos, la presencia de antimieloperoxidasa ANCA (p-ANCA) asociado con mayor fibrosis intersticial, atrofia tubular y esclerosis glomerular17, de esta forma a mayor glomeruloesclerosis y fibrosis intersticial en biopsia renal, peor pronóstico renal a largo plazo, aunque reciba tratamiento inmunosupresor intensivo incluyendo plasmaféresis15.
En los pacientes que atendimos, consideramos que su evolución a 12 meses fue el resultado de una coexistencia de varios de estos factores: inflamación tubulointersticial mononuclear en su mayoría (87%), alta necrosis fibrinoide y arteriolopatía moderada a severa (83%), alta proporción de glomérulos con proliferación extracapilar, así como esclerosis segmentaria y global (57%) y fibrosis intersticial de al menos un 35% del tejido renal. Esto sin tomar en cuenta criterios clínicos, como la edad del paciente, comorbilidades subyacentes y el tiempo de evolución de la glomerulonefritis sin recibir intervención inmunosupresora; esta última, consideramos que jugó un papel importante en la perpetuación del estado inflamatorio del tejido renal, ocasionando esclerosis glomerular y fibrosis intersticial extensa, dejando poco o nada de tejido viable al momento de la intervención, lo que obviamente reduce la sobrevida renal aunque se empleen medidas novedosas como anticuerpos monoclonales y plasmaféresis.
Existe un estudio en el que se establecen criterios histomanifestacioneslógicos y clínicos como predictores de mala evolución hacia la terapia dialítica definitiva al momento del ingreso y a los 12 meses, así en análisis multivariado existe una correlación negativa a presentar mayor deterioro de la función renal con la edad del paciente, atrofia tubular, crescencias fibrosas, lesión tubulointersticial aguda y crónica; del mismo modo, se encontró una asociación con mejor función renal e independencia dialítica a 12 meses entre mayor porcentaje de glomérulos normales encontrados en la biopsia renal y el uso de plasmaféresis como opción de tratamiento18.
Trasplante renal en vasculitis ANCA
Con el inicio de la falla renal y la terapia dialítica muchas glomerulonefritis reducen su actividad al punto de desaparecer, sin embargo, algunas continúan con actividad y pasa desapercibida incrementando la mortalidad asociada, por lo que debe monitorearse a los pacientes19. Una vez trasplantados, los pacientes que se mantienen con un régimen a base de prednisona, micofenolato y ciclosporina o tacrolimus, tienen una evolución satisfactoria, con buen pronóstico y tasas altas de sobrevida para el paciente y sus injertos. La recurrencia de la glomerulonefritis activa en el injerto renal es de 15 a 37% y responden bien a terapia con ciclofosfamida y otras terapias más intensivas2,13.
En nuestra serie, 3 de los pacientes fueron sometidos con éxito a trasplante renal, sin complicaciones posteriores de recidiva en más de 4 años de evolución con sus injertos, importante mencionar que otros pacientes están actualmente en protocolo de trasplante, pero no ha sido posible concretar la cirugía por motivos ajenos a la etiología de la enfermedad renal crónica o consenso del equipo de trasplante, teniendo presente que el antecedente de la vasculitis no constituye una contraindicación para el mismo.
Conclusiones
La vasculitis renal ANCA asociada tiene un pobre pronóstico a corto plazo, cuya sobrevida está íntimamente relacionada al tiempo de evolución de la actividad de la enfermedad y su intervención inmunosupresora oportuna, por lo que la detección temprana mediante búsqueda intencionada ante la mínima sospecha es trascendental en la mejora del pronóstico de este tipo de pacientes.
Responsabilidades éticas
Protección de personas y animales. Los autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datos. Los autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informado. Los autores declaran que en este artículo no aparecen datos de pacientes.

