











 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkIntroduction
Pompe disease (glycogen storage disease type II, OMIM 232300) is a rare disease of autosomal recessive inheritance, initially described in 1932 by the Dutch pathologist Johannes C. Pompe.1,2It is caused by the genetic deficiency of the lysosomal enzyme acid a-glucosidase (AAG), which leads to abnormal accumulation of glycogen in the cells, with dysfunction of multiple organs, but mainly the muscles.3,4 The accumulation of glycogen in the striated muscle causes muscle dysfunction in patients, which is typically manifested by muscle weakness of proximal predominance, being a differential diagnosis of other myopathies. The muscle commitment can extend to the respiratory muscles generating diaphragmatic paralysis, alveolar hypoventilation and, in some cases, respiratory failure and death.3,5,6Cardiac accumulation with manifestations such as cardiac hypertrophy and cardiac rhythm disorders has also been documented.3,7,8The disease has an infantile presentation but there is also a late presentation (after one year of age), which is a diagnostic challenge.4,7,8In patients with the classical infantile form the disease manifests itself in the first months of life with hypertrophic cardiomyopathy, significant hypotonia and skeletal muscle weakness and, if they are not treated, they typically die from heart failure during the first year of life.6,8Patients with the late-onset form have a milder phenotype, which typically consists of proximal skeletal muscle weakness with slow progressive myopathy, but they rarely have cardiac affectation.4,6,7,9The early onset of the disease results from a complete or almost complete deficiency of the functional AAG protein, while late-onset patients maintain some residual enzyme activity.2 The late form, and especially when it occurs after the fifth decade, is not common.6
Clinical case
A 60-year-old male patient, who had an established diagnosis of an autoimmune myopathy of polymyositis type and met 3 of 4 criteria of Bohan and Peter, supported by progressive muscle weakness, electromyographic alterations with a myopathic pattern and elevation of muscle enzymes, was managed with prednisolone 30 mg daily, azathioprine 75 mg daily and physical therapy. The patient showed a poor response to the treatment established, and therefore he decided to consult.
The clinical examination showed significant atrophy in the anterior and posterior muscles of the arms, as well as in the thighs and hips. The muscle strength in the upper limbs was decreased: proximal 3/5 and distal 4/5. Weakness was also observed in the lower limbs: 2/5 proximal and 2/5 distal, in addition to patellar hyporreflexia. There was no sensory deficit. The Gowers' sign was positive. The rest of the clinical examination and the vital parameters were normal.
Due to the patient's poor response to the treatment received, it was decided to reconsider the diagnosis and perform new studies (Table 1), which included a metabolic profile that was normal, an electromyography of the four extremities and a muscle biopsy. The electromyography was abnormal, showing alterations compatible with progressive muscular dystrophy and there were abundant signs of hyperexcitability of the cell membrane, characteristic of inflammatory myopathies. The muscle biopsy of the distal third of the biceps of the left arm showed loss of muscle fibers with replacement by adipose tissue. When immunohistochemical studies were performed, no inflammation was observed and infiltrates indicating an inflammatory myopathy were not found. Periodic acid Schiff (PAS) stains were not performed.

Given the diagnostic uncertainty, a screening for Pompe disease was carried out with a blood spot on filter paper, which was altered, observing a high neutral/inhibited α-glucosidase ratio and a high percentage of inhibition of the AAG. Taking into account the positive screening test, an enzymatic study on leukocytes was carried out, with a neutral/inhibited ratio that was also altered, thus confirming an alteration of the AAG. (Table 2).
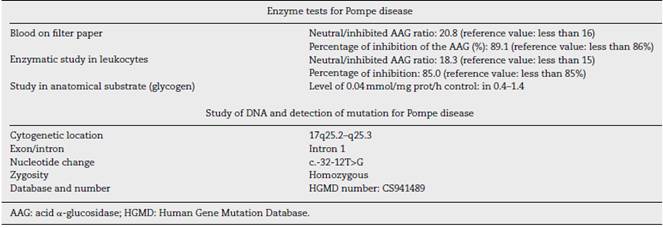
Finally, it was decided to perform a study on anatomical substrate (glycogen), which showed an abnormally low level, with which the diagnosis of adult-onset Pompe disease was confirmed. In addition, it was carried out a genetic study that showed c.-32-12T>G alteration in the homozygous state of the AAG gene intron 1 (Table 2). After the diagnosis, management with enzyme replacement was carried out with recombinant human α-glucosidase, with doses of 20 mg/kg by slow intravenous perfusion every 15 days. After the enzyme replacement therapy was started, not only stability of the disease was observed, but also improvement of weakness at 3 months of follow-up, showing strength of 3/5 bilaterally in the lower limbs. The patient continues on clinical follow-up by rheumatology.
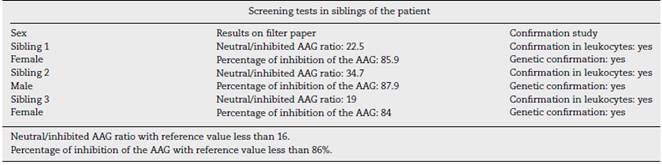
Taking into account the genetic nature of the disease, screening of 10 siblings of the patient was done, being able to detect alteration in the enzymatic function in 3 of them (Table 3). A confirmatory study with a leukocyte test and a genetic study which confirmed the alteration and the mutation of the AAG were also carried out; however, at the time of the exam none of them showed symptoms.
Discussion
Pompe disease is considered a rare condition, with an incidence between 1:14,000 and 1:300,000, depending on the geographic area studied, being the highest incidence of the infantile form among Chinese people, while the adult late-onset form has the highest incidence in the Netherlands.7,10The diagnosis is difficult and is usually delayed between 4 and 30 years after the onset of the symptoms.3 The age of onset can range from <1 to 52 years and the age of diagnosis can vary from <1 to 78 years.9 In the case presented, the patient was diagnosed with a polymyositis, but he did not had a good response to the management established, and for this reason, diagnostic alternatives were sought, which included: congenital, inflammatory (other than polymyositis, inclusion bodies, dermatomyositis), endocrine, mitochondrial, and metabolic (McArdle disease, carnitine palmitoyltransferase type II deficiency, Pompe disease) myopathies, muscular dystrophies (mainly Duchenne dystrophy, Becker dystrophy and limb-girdle dystrophy).8,11Other differential diagnoses that can be included are the upper and lower motor neuron diseases, which may also occur with muscle atrophy and weakness.12After a positive screening test with blood spot on filter paper, it was possible to focus on a deficit of AAG.
The clinical manifestations of adult-onset Pompe disease are varied, being the most frequent proximal muscle weakness, trunk muscle weakness, intolerance to exercise, difficulty breathing due to respiratory muscle weakness (in up to 80%), ineffective cough, difficulty walking and difficulty in getting up from a squatting position (known as Gowers' sign). However, it can have manifestations in multiple organs including the heart (hypertrophic cardiomyopathy, supraventricular tachycardia and Wolff-Parkinson-White syndrome), the nervous system (small fiber neuropathy, hearing loss), the vascular system (cerebral aneurysms, basilar dolichoectasia), the osseous system (osteopenia, osteoporosis, scoliosis) and the gastrointestinal system (chronic diarrhea, fecal incontinence).2,3,6,7,11From the biochemical point of view, a significant percentage of patients can present with elevation of creatine phosphokinase (CPK) and other muscle enzymes, which masks the diagnosis and patients can be misdiagnosed with inflammatory myopathies for several years.13
The diagnostic standard for Pompe disease is the demonstration of the deficiency of the enzymatic activity of the AAG.4,7,8,14The biochemical measurement can be performed on samples of cultured skin fibroblasts, muscle tissue biopsy or blood samples on filter paper, purified lymphocytes and mixed leukocytes4,7,10,15Other forms of diagnosis that can be established are through a muscle biopsy with positive PAS staining, taking into account that the muscle glycogen content increases up to 10-fold above the normal value in the infantile Pompe disease and to a lesser extent in late-onset patients.4,6,7
The gene that encodes the AAG is located on chromosome 17q25.3 and its mutation is associated with the autosomal recessive Pompe disease.2,6,8,9 More than 350 mutations of the disease have been described.2,9,16 Among the recurrent mutations in the cases of infantile onset there is a deletion of ∆525T, which is observed in 9% of cases in the United States and in 34% of the Dutch cases, and the mutation of the splice site of IVS1 (-13T->G) represents approximately 50% of the cases of late-onset in Caucasian patients.7,8 In our case, the intronic mutation c.-32-12T>G in the homozygous state of the AAG gene was documented, which corresponds to the most frequent pathogenic mutation for late-onset Pompe disease. Although this mutation was documented for the first time many years ago (in 1994 by Huie et al.), the mechanism by which it affects the encoding of the AAG is not clear so far.17
Once a patient is diagnosed with Pompe disease, it is ideal to have a basic study of the cardiopulmonary structure and function, being indicated in the management guidelines the performance of a chest X-ray, an electrocardiogram and an echocardiogram at the first visit. In the adult forms it is ideally recommended to carry out pulmonary function tests with spirometry, lung volumes with measurement of the maximum expiratory pressure and the maximum inspiratory pressure. It is also important to provide patients with an integral rehabilitation plan that allows to preserve the motor an physiological function, prevent or minimize the secondary complications and maximize the benefits of the enzyme replacement therapy (ERT).6,7,11
ERT was approved in 2007 for patients with early-onset Pompe disease and in 2010 for the late presentation.8,11This therapy has shown benefits, especially when it is started early.2,4,10,18The therapy with the recombinant human AAG enzyme (Myozyme®) has a standard dose of 15-20 mg/kg every 15 days in continuous infusion for 4h.2,11
Since it is an autosomal recessive disease, the probability of transmission of the disease from a parent to a child is approximately 25%, therefore, genetic counseling should be carried out.7,8,11 The active search for the disease among siblings is also justified, as in this case, where it was possible to determine the enzymatic alteration in 3 of 10 siblings; however, they may be asymptomatic or slightly symptomatic due to intrafamilial clinical variability.8,11 Currently, the evidence is insufficient to treat the presymptomatic disease or for the management of the mutation carriers.14
Conclusion
Late-onset Pompe disease is a rare condition, with low suspicion, that may go unnoticed if it is not taken into account within the differential diagnoses of inflammatory myopathies, since it may present similar clinical manifestations with proximal muscle weakness and elevated muscle enzymes. Its early detection is important to initiate an ERT that allows to stop the clinical deterioration and to avoid the derived complications improving the quality of life of the patients.

