










Servicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Colombiana de Reumatología
versión impresa ISSN 0121-8123
Rev.Colomb.Reumatol. vol.26 no.4 Bogotá oct./dic. 2019 Epub 01-Nov-2020
Reporte de casos
Hemorragia severa secundaria a deficiencia adquirida de factores VIII y XI de la coagulación relacionada con lupus eritematoso sistémico: reporte de caso
a Grupo de Investigación en Medicina Interna, Facultad de Medicina, Universidad Tecnológica de Pereira, Pereira, Risaralda, Colombia
b Servicio de Reumatología, Clínica Los Rosales, Pereira, Risaralda, Colombia
La ocurrencia simultánea de anticoagulante lúpico e inhibidores circulantes contra los factores de la coagulación es infrecuente en los pacientes con enfermedad autoinmune, y está relacionada con eventos hemorrágicos. El abordaje inicial requiere una adecuada interpretación de los tiempos de coagulación y prueba de mezcla con plasma para alcanzar el diagnóstico definitivo. Se reporta el caso de una paciente con lupus eritematoso sistémico y síndrome de Sjögren, quien se presentó con trastorno hemorrágico amenazante de la vida ocasionado por inhibidores circulantes contra los factores VIII y XI de la coagulación en coexistencia con anticoagulante lúpico. El tratamiento de erradicación de los inhibidores se realizó con rituximab, con buenos resultados.
Palabras clave: Deficiencia del factor VIII; Deficiencia del factor XI; Inhibidor de coagulación del lupus; Lupus eritematoso sistémico; Inhibidores de factor de coagulación sanguínea; Rituximab
In patients with autoimmune diseases, the simultaneous occurrence of lupus anticoagulant and blood coagulation factors inhibitors is infrequent and is associated with hemorrhagic events. In these cases, the initial approach requires a thorough interpretation of coagulation laboratory tests and mixing studies to reach a definitive diagnosis. We report the case of a patient with systemic lupus erythematosus and Sjögren's syndrome who presented with hemorrhagic diathesis caused by circulating inhibitors against factors VIII and XI coexisting with lupus anticoagulant. The inhibitors eradication was made with rituximab, achieving good results.
Keywords: Factor VIII deficiency; Factor XI deficiency; Lupus coagulation inhibitor; Systemic lupus erythematosus; Blood coagulation factor inhibitors; Rituximab
Introducción
El lupus eritematoso sistémico (LES) es el prototipo de enfermedad inflamatoria crónica con desregulación del sistema inmune y producción de autoanticuerpos1. La presencia de anticuerpos antifosfolípidos en el LES es frecuente (15-34% para anticoagulante lúpico [AL]) y se asocia con el desarrollo de eventos trombóticos2. En menor proporción, anticuerpos dirigidos contra factores de la coagulación (II, VIII, IX, XI, XII y XIII) han sido reportados3. Estos anticuerpos alteran la función o promueven la depuración rápida de los factores de la coagulación y, por consiguiente, se manifiestan con trastornos hemorrágicos4. En pacientes con enfermedad autoinmune, la ocurrencia simultánea de AL e inhibidores de factores de la coagulación es infrecuente.
Caso clínico
Mujer de 65 anos diagnosticada 6 meses antes con LES de inicio tardío, según criterios SLICC 2012, sustentado en: alopecia, aftas orales, fotosensibilidad, consumo de complemento C3, trombocitopenia < 100.000 µl, leucopenia < 4.000 µl, Coombs directo positivo ++, ANA (1/160 patrón homogéneo), anti-Sm 14,74 (positivo); con AL negativo al momento del diagnóstico. Además, con síndrome de Sjögren secundario por presencia de xerostomía, xeroftalmia, xerodermia, anti-Ro 26,89 (positivo), anti-La 51,2 (positivo), en manejo con prednisolona 5 mg/día, azatioprina 50 mg/día e hidroxicloroquina 200 mg/día.
Ingresó a hospitalización por dolor abdominal y hematomas en extremidades. Sin antecedentes familiares o personales de diátesis hemorrágica; no había historia de trauma y fue negado el consumo de medicamentos o productos herbales con acción anticoagulante. Al examen físico se encontró en regular condición general, con palidez mucocutánea, hipotensión arterial y taquicardia. Se observó una lesión equimótica en región posterior de faringe, hematomas en brazo derecho, antebrazo izquierdo, región inguinal derecha y muslo ipsilateral (fig. 1). Los estudios de laboratorio revelaron leucopenia, anemia severa, trombocitopenia moderada y linfopenia. El tiempo de tromboplastina parcial activada (TTPa) fue prolongado en 190 s (normal: 20-35 s), con tiempo de protrombina (TP) en 10,3 s (normal: 9-13 s) y prueba de mezcla con plasma normal que no corrigió. Mediante estudio ecográfico se evidenció hemartrosis en hombro derecho, hematoma en vasto lateral y bíceps femoral derecho, hematoma retroperitoneal adyacente al músculo psoas derecho y trombosis venosa de la vena cefálica, cervical y braquial. No hubo signos de isquemia crítica en extremidades que hicieran necesaria una intervención quirúrgica urgente.
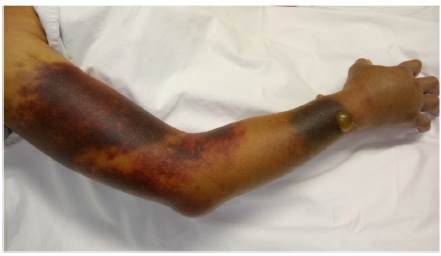
Estudios adicionales revelaron la presencia de AL positivo e inhibidores circulantes contra los factores VIII y XI de la coagulación, así como deficiencia cuantitativa de los niveles de estos (tabla 1).

El manejo inicial se realizó con soporte transfusional de concentrado globular y plasma fresco congelado en combinación con pulsos de metilprednisolona durante 3 días consecutivos, seguido de prednisolona oral a dosis de 1mg/kg/día y ácido tranexámico, sin obtener respuesta, no se contaba con la opción terapéutica de factor VII activado recombinante de urgencia; dada la leucopenia y linfopenia severa, por perfil de seguridad no se decidió aplicar ciclofosfamida, por lo que se indicó manejo con rituximab a dosis de 1 g intravenoso en esquema semana 0 y a los 15 días, durante la hospitalización, para erradicación de los inhibidores, obteniéndose una buena evolución clínica y corrección de los tiempos de coagulación, después de la primera aplicación, la paciente continuó en controles con hematología trimestral elevando niveles de los factores de coagulación, con reaplicación del rituximab a los 6 meses y estudios negativos para neoplasia oculta, sin necesidad de reingreso hospitalario por infección o sangrado después de 12 meses.
Discusión
Describimos el caso de una paciente con LES y síndrome de Sjögren que ingresó por trastorno hemorrágico en quien los laboratorios iniciales revelaron un TTPa elevado con TP normal y prueba de mezcla con plasma normal que no corrigió, orientando hacia la presencia de inhibidores adquiridos de la coagulación ya sea AL o inhibidores circulantes contra los factores de coagulación4,5. Para ayudar a aclarar la etiología, la presentación clínica resulta útil porque, aunque el AL in vitro aumenta el TTPa, in vivo se relaciona con eventos trombóticos venosos y arteriales6; mientras que los inhibidores circulantes contra los factores de coagulación se asocian con eventos hemorrágicos7. En adición a la clínica, se requiere confirmación definitiva por el laboratorio con medición del AL, niveles de los factores de la coagulación e inhibidores circulantes4,5. En este caso, el resultado confirmatorio de las pruebas de laboratorio demostró la presencia de inhibidores circulantes contra los factores VIII y XI en títulos muy altos, así como disminución cuantitativa de los niveles de los respectivos factores.
La aparición de inhibidores contra los factores de la coagulación en pacientes no hemofílicos es poco común, con una incidencia aproximada de 2 personas por millón8, con una edad media de aparición de 64 anos e incidencia varón/mujer similar9. A diferencia de la forma congénita donde las hemorragias articulares (hemartrosis) son la característica principal, estas son poco comunes en la hemofilia adquirida, manifestándose principalmente con hemorragias cutáneas y en tejidos blandos8. Cerca del 50% de los casos se encuentran relacionados con una condición clínica subyacente (desórdenes autoinmunes, enfermedades linfoproliferativas, tumores sólidos, medicamentos, embarazo y posparto) 9, siendo el LES la más frecuente de las causas reumatológicas10. Dada la desregulación inmune que ocurre en el paciente con LES, auto-anticuerpos policlonales principalmente de tipo IgG 1 y 4, son dirigidos contra los dominios A2 o C2 del factor VIII neutralizando su función o promoviendo una rápida depuración de este en la circulación4,11. Aún en presencia de títulos altos de inhibidores como los vistos en este caso, se puede encontrar actividad residual cuantificable de los factores de la coagulación, evidenciando una interacción compleja en la cinética entre inhibidor y factor, que es diferente a lo que ocurre en la hemofilia congénita. Por ende, la gravedad del sangrado se correlaciona pobremente con los niveles del factor8,11.
Un factor agravante en esta paciente, fue la deficiencia de factor XI, la cual ha sido raramente descrita en pacientes con LES causando sangrado amenazante de la vida12. La deficiencia combinada de los factores VIII y XI en forma adquirida, aunque es presumible que compartan la misma patogenia, no ha sido reportada previamente.
La ocurrencia simultánea de inhibidores circulantes de la coagulación con AL es inusual. Existen reportes de caso desde 1993 en asociación con enfermedades del tejido conectivo y trastornos mieloproliferativos13, ocasionando trastornos hemorrágicos principalmente, pero también eventos trombóticos como fue visto en esta paciente. Se resalta el hecho que la manifestación hematológica en esta paciente ocurrió en ausencia de otras manifestaciones clínicas de actividad de la enfermedad.
El objetivo terapéutico se basó en controlar el sangrado y erradicar el inhibidor. El control del sangrado oportuno disminuye la morbimortalidad. Los agentes puente como el factor VIIa recombinante y el concentrado de complejo protrombínico activado son agentes de primera línea5, siendo menos útil el plasma fresco congelado por su bajo contenido del factor, sin embargo, este último fue utilizado como único recurso por la falta de disponibilidad de los agentes puente en nuestro hospital. El riesgo de trombosis debe tenerse en cuenta con la utilización de agentes puente, aunque este no contraindica su uso dado que el beneficio del control del sangrado sobrepasa los riesgos. Dado el sangrado amenazante de la vida y la ausencia de utilización de agentes puente de la coagulación fue considerado no utilizar terapia anticoagulante para el manejo de la trombosis de extremidad superior.
Para la erradicación de los inhibidores circulantes contra los factores de coagulación, además del control de la enfermedad de base, se han utilizado los medicamentos inmunosupresores (ciclofosfamida, azatioprina y rituximab), glucocorticoides, gammaglobulina intravenosa y plasmaféresis, con resultados satisfactorios. La discusión se ha centrado sobre el régimen más efectivo. A pesar de que diferentes estudios muestran que los pacientes tratados en primera línea con una combinación de esteroides y ciclofosfamida, fue más probable que aseguraran remisión completa que los tratados con esteroides solos, el resultado final en términos de supervivencia y remisión sostenida es el mismo5. Rituximab es una opción terapéutica cada vez más empleada y con resultados similares a ciclofosfamida en cuanto a eficacia, pero con mejor perfil de seguridad. En este caso el uso de rituximab resultó en remisión completa de la enfermedad con normalización de los factores de coagulación, erradicación del inhibidor y reducción de la inmunosupresión.
Conclusión
El LES está asociado con la producción de inhibidores circulantes contra factores de la coagulación, entre ellos, los factores VIII y XI ocasionando trastornos hemorrágicos. Si bien esta es una asociación poco frecuente, debe ser estudiada en pacientes con LES con pruebas de coagulación anormales, aun en ausencia de otras manifestaciones de la enfermedad, con el fin de mejorar la calidad de vida y disminuir la tasa de morbimortalidad de estas enfermedades.
REFERENCIAS
1. Kaul A, Gordon C, Crow MK, Touma Z, Urowitz MB, van Vollenhoven R, et al. Systemic lupus erythematosus. Nat Rev Dis Primers. 2016;2:16039. [ Links ]
2. Schreiber K, Sciascia S, de Groot PG, Devreese K, Jacobsen S, Ruiz-Irastorza G, et al. Antiphospholipid syndrome. Nat Rev Dis Primers. 2018;4:17103. [ Links ]
3. Monrad SU, Kaplan MJ. CHAPTER 48 - Cellular Hematology A2 - Lahita, Robert G. Systemic Lupus Erythematosus. 5th ed. San Diego: Academic Press; 2011. p. 905-20. [ Links ]
4. Cugno M, Gualtierotti R, Tedeschi A, Meroni PL. Autoantibodies to coagulation factors: from pathophysiology to diagnosis and therapy. Autoimmun Rev. 2014;13(1):40-8. [ Links ]
5. Collins PW, Chalmers E, Hart D, Jennings I, Liesner R, Rangarajan S, et al. Diagnosis and management of acquired coagulation inhibitors: a guideline from UKHCDO. Br J Haematol. 2013;162(6):758-73. [ Links ]
6. Chighizola CB, Raschi E, Banzato A, Borghi MO, Pengo V, Meroni PL. The challenges of lupus anticoagulants. Expert Rev Hematol. 2016;9(4):389-400. [ Links ]
7. Seethala S, Collins NP Jr, Comerci G Jr. An unusual etiology for elevation of activated partial thromboplastin time (aPTT) in SLE: acquired hemophilia and lupus anticoagulant. Case Rep Hematol. 2013;2013:521785. [ Links ]
8. Giangrande P. Adquired hemophilia: revised edition. Treatment Hemophilia. 2012;38:1-7. [ Links ]
9. Delgado J, Jiménez-Yuste V, Hernández-Navarro F, Villar A. Acquired haemophilia: review and meta-analysis focused on therapy and prognostic factors. Br J Haematol. 2003;121(1):21-35. [ Links ]
10. O'Connor CR. Systematic review of the presentation of coagulation factor VIII inhibitors in rheumatic diseases: a potential cause of life-threatening hemorrhage. Semin Arthritis Rheum. 2015;44(6):695-709. [ Links ]
11. Collins PW, Percy CL. Advances in the understanding of acquired haemophilia A: implications for clinical practice. Br J Haematol. 2010;148(2):183-94. [ Links ]
12. Bortoli R, Monticielo OA, Chakr RM, Palominos PE, Rohsig LM, Kohem CL, et al. Acquired factor XI inhibitor in systemic lupus erythematosuscas report and literature review. Semin Arthritis Rheum. 2009;39(1):61-5. [ Links ]
13. Gupta D, Chatterjee T, Sharma A, Ganguli P, Das S, Sharma S. Rare case of acquired haemophilia and lupus anticoagulant. Indian J Hematol Blood Transfus. 2014;30(3):197-200. [ Links ]
Financiación Los autores declaran no haber recibido financiación para la realización de este trabajo.
Recibido: 09 de Octubre de 2018; Aprobado: 07 de Febrero de 2019; : 09 de Diciembre de 2019
 This is an open-access article distributed under the terms of the Creative Commons Attribution License
This is an open-access article distributed under the terms of the Creative Commons Attribution License
