











 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroduction
Systemic sclerosis (SSc) is a systemic connective tissue-associated autoimmune disease, characterized by the development of microvascular injury, generalized fibrosis with multi-organ compromise, and inadequate regulation of the immune system. The worldwide prevalence of the disease is variable depending on the population studied. The disease is less frequent in populations from northern Europe and Japan, while in southern Europe, North America and Australia it is more prevalent. Similarly to other connective tissue-associated autoimmune diseases, it is more frequent in women. However, the disease has a worse prognosis in men, as it is associated with higher risk of diffuse cutaneous compromise and mortality.1
Based on the extension of cutaneous compromise, the disease is classified into: limited SSc, diffuse SSc, and SSc sine scleroderma. Additionally, it may present as an overlap syndrome, in which clinical characteristics from other connective tissue-associated autoimmune diseases, such as systemic lupus erythematosus, are present. Considering the natural history of the disease, it has been determined that the extension of cutaneous compromise and the rate of progression are markers of possible internal organ compromise including the esophagus, lungs, kidneys, etc.1
Pulmonary compromise is one of the most frequent complications, and it has high impact on morbidity and mortality. The main pulmonary complications that could be developed in these patients are fibrosis (associated with the tendency of the disease to generate aberrant wound healing process) and pulmonary hypertension (associated with proliferative vasculopathy).2 The pulmonary complications have replaced renal crisis as the main cause of mortality in patients with SSc.4
Systemic sclerosis-associated pulmonary hypertension
Systemic sclerosis-associated pulmonary hypertension (SSc-PHT) is a frequent and severe complication of these patients. It affects approximately 10% of patients, and it is associated with a poor survival since diagnosis, which is approximately 3 years.3
Pulmonary hypertension was defined as a mean pulmonary artery pressure above 25 mmHg, and a pulmonary artery wedge pressure below 15 mmHg, measured by right heart catheterization, in absence of interstitial lung disease.4
The pathophysiology of the disease is very complex, and mainly covers the development of vascular lesions secondary to injury of the pulmonary endothelium and endothelial dysfunction, producing an aberrant fibrosis and an inadequate vascular repair, resulting in an obliterating vasculopathy. The arterioles present an important thickening due to increased muscle tissue and perivascular inflammatory changes associated with an inadequate function of monocytes/macrophages. Additionally, the presence of endothelial dysfunction and oxidative stress contributes greatly to the pathogenesis of the disease.2
Pulmonary hypertension is classified by the World Health Organization into 5 categories based on the clinical and pathophysiological characteristics:
I. Pre-capillary pulmonary arterial hypertension I'. Pulmonary veno-occlusive disease
II. Post-capillary pulmonary hypertension
III. Pulmonary hypertension due to lung fibrosis or hypoxia
IV. Thromboembolic pulmonary hypertension
V. Uncommon and multifactorial pulmonary hypertension
In patients with SSc, the most frequent form is type I, although they could also present mixed forms of the disease.2
The phenotypic definition of pulmonary hypertension that affects each patient continues to be a difficult process due to the tendency to present multiple etiologies of pulmonary hypertension in the context of SSc. Comorbidities of pulmonary hypertension and pulmonary fibrosis are present in 25-50% of patients, especially in those with diffuse disease. These patients have a worse prognosis and poor response to treatment. Pulmonary veno-occlusive disease is characterized by a diffuse obstruction of small pulmonary veins, and it may be found in 61% of patients with SSc-PHT. This condition is also associated with a lower survival and an increased risk of pulmonary edema after initiating the treatment. Additionally, the cardiac compromise given by myocardial fibrosis could be a cause of SSc-PHT, especially type II, by generating left ventricular dysfunction.3
Pharmacological treatment of SSc-PHT has presented great advances in recent years, especially with the development of new oral medications and the benefit of combined therapies with different mechanisms of action.2 The treatments are based on the induction of vasodilation, and for that purpose, the following mechanisms of action have been used: endothelin-1 receptor antagonists (Bosentan, Ambrisentan, Macitentan, etc.), Phosphodiesterase inhibitors (Sildenafil, Tadalafil), guanylate cyclase inhibitors (Riociguat), and prostacyclin analogs (Epoprostenol, Treprostinil).1
The aim of this systematic literature review is to assess the efficacy of the different pharmacological treatments available for the management of SSc-PHT.
Materials and methods
Types of studies
Randomized controlled clinical trials, non-randomized controlled clinical trials, and observational studies including cohort and case-control studies.
Observational studies without comparison group, studies that did not assess the proposed outcomes, duplicated publications, studies on-course, and studies without enough information to extract outcomes of interest for patients with SSc were excluded.
Population
Studies with adult patients (at least 18 years of age) with diagnosis of SSc-PHT using any of the pharmacological treatments available were included.
Types of intervention
Studies in which some of the treatments available for SSc-PHT (prostacyclin analogs, endothelin receptor antagonists, phosphodiesterase inhibitors and soluble guanylate cyclase stimulators) were compared against placebo or a medication from another group used to treat SSc-PHT.
Outcomes
The primary outcomes included in the study were exercise capacity measured with the 6-minute walk test, functionality measured with WHO functional class or NYHA functional classification, Borg dyspnea score. The secondary outcomes included cardiopulmonary hemodynamic measurements, NT-proBNP levels, mortality and adverse effects.
Search methods
The search for publications was made in the following scientific electronic databases: CENTRAL (Cochrane Central Register of Controlled Trials), MEDLINE, and EMBASE. The search was made between January 1966 and April 2019.
The search terms included were: (("Hypertension, Pulmonary"[Mesh] OR "pulmonary hypertension" OR "pulmonary arterial hypertension") AND ("Scleroderma, Systemic"[Mesh] OR "systemic sclerosis")) AND ((treatment OR "Therapeutics"[Mesh]) OR (Bosentan OR Ambrisentan OR Macitentan OR Sitaxsentan OR Sildenafil OR Tadalafil OR Vardenafil OR Riociguat OR Selexipag OR Epoprostenol OR Iloprost OR Treprostinil OR Beraprost)).
Selection of studies
Selected studies were assessed by two researchers. The following data was obtained: publication details (title, first author, publication date), study design, details of the study participants (number of patients included, demographic characteristics), type of intervention, type of comparator, follow-up time, and outcomes with their corresponding effect measurement.
Quality and risk of bias assessment
The methodological quality of the studies was assessed based on the type of study design, following the Cochran Handbook for Systematic Reviews of Interventions,5 including the risk of bias. In cohort and case-control studies, the Scottish Intercollegiate Guidelines Network (SIGN) checklist was applied for the methodological assessment.6
Measurement of treatment effects
For the measurement of pharmacological treatment effects, a descriptive and exploratory analysis for every quantitative variable was presented by central tendency and dispersion measurements, and by frequency measurements for qualitative variables, including descriptive graphics. Finally, based on the data available, a meta-analysis was developed when it was possible.
Results
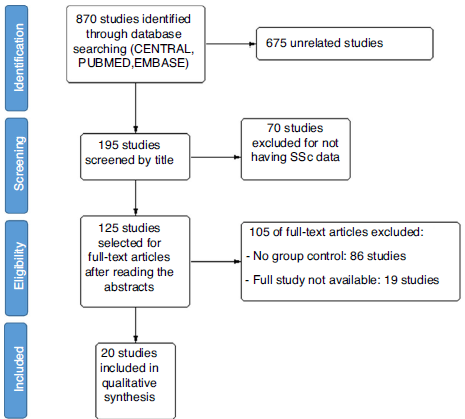
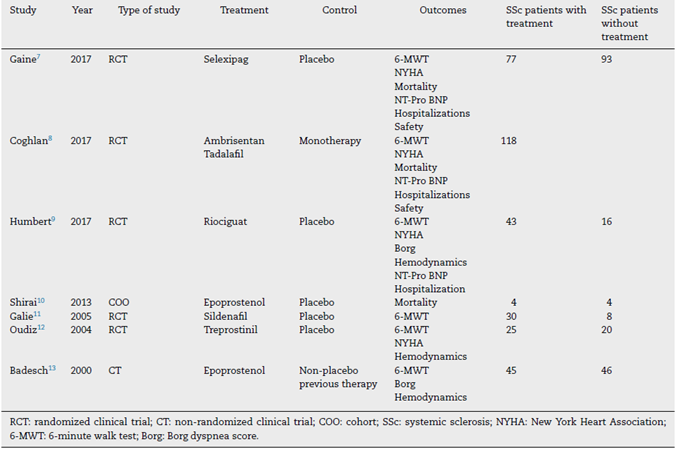
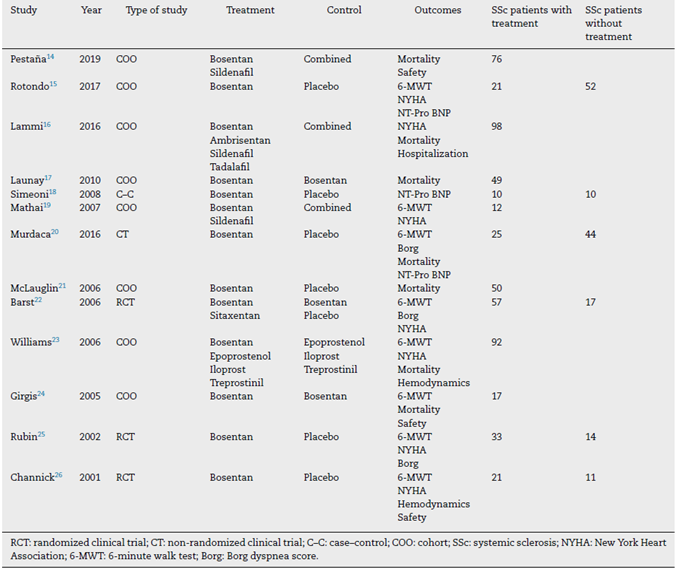
A total of 870 studies were found during the initial search. Of these, 195 studies were selected based on the title; then 120 studies were selected based on abstracts for full reading. Finally, 20 studies were included, as they fulfilled the inclusion criteria (see Fig. 1). The definition of arterial pulmonary hypertension had been changed since 2019, lowering the normal value of median pulmonary pressure to 20 mmHg, however, all of the studies used the previous cut-off point of 25 mmHg. Of the 20 studies, 8 were randomized clinical trials, 2 were non-randomized clinical trials, 9 cohorts, and 1 case-control study. Of these, 13 studies included Bosentan as monotherapy or in combination. A summary of the characteristics of the studies is described in Tables 1 and 2.
After this search and data analysis, an active search was done specifically for Macitentan, Vardenafil and Beraprost, without finding any additional reference.
The quality of evidence assessment found that in general, the clinical trials have an overall adequate quality. The risk of bias in the clinical trials is presented in Figs. 2 and 3. The study with more limitations was the one conducted by Badesh and colleagues13 that uses Epoprostenol. It is important that the other study using this medication is of low quality,10 as it is going to be presented later. The most frequent biases in the studies were related to the blinding of participants, personnel and researchers.
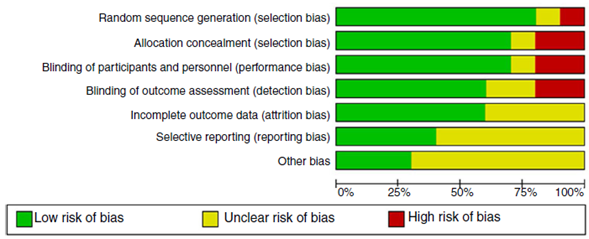
Fig. 2 Risk of bias graph: reviews authors'judgments about each risk of bias item presented as percentages across all included clinical trial studies.
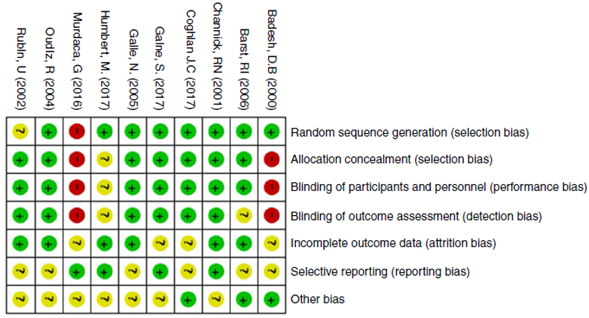
Fig. 3 Risk of bias summary: reviews authors' judgments about each risk of bias item for each included clinical trial study.
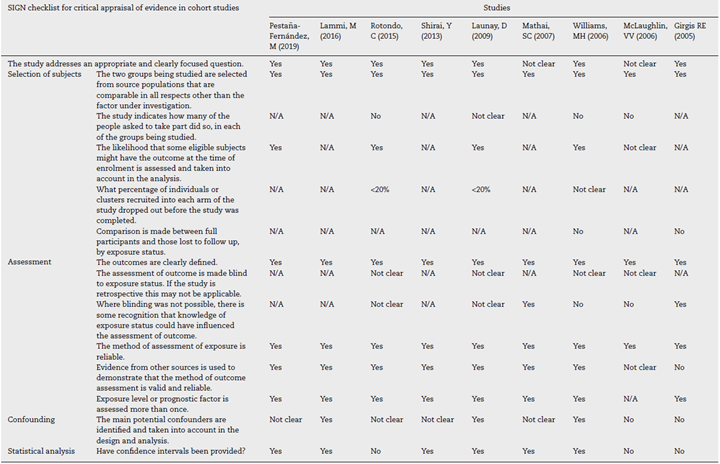
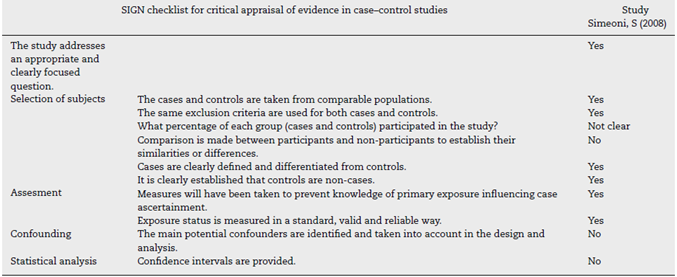
The quality assessment of cohort and case-control studies using the SIGN checklist specific for each type of study is presented in Table 3 and Table 4, respectively. In general, the quality of the studies was not the best, as several biases may be present, according to the checklist. The most frequent biases were the lack of data regarding desertion of participants, data analysis of deserting participants, and blinding during outcome analysis.
The studies that did not include Bosentan as treatment are mostly clinical trials. A total of 342 patients with SSc-PHT with treatment, and 187 patients with placebo were included. Except for two studies, all of them used different medications. The two studies that used Epoprostenol as treatment,10,13 could not be used in posterior analysis due to the type of study and the limited amount of patients included. Given these characteristics of the studies, it was not possible to do a meta-analysis in the group that did not use Bosentan.
Regarding the results in patients with SSc, Xelexipag7 reduced the risk of events based on a compound outcome (including mortality, changes in functional class, hospitalizations and adverse events, requirement of lung transplants, requirement of balloon atrial septostomy, and use of intravenous prostanoids), compared to placebo (HR: 0.59; 95% CI = 0.41-0.85). Selected patients in the analysis of the study were part of the GRIPHON trial, and they were grouped with those with an autoimmune etiology of the PHT, including patients with SSc, systemic lupus erythematosus, mixed connective tissue disease, and non-differentiated.
Riociguat9 at a dose of 2.5 mg/day, improved the 6-minute walk distance test (18±51 m), compared to placebo (-8±110 m) after 12 months during the PATENT-1 trial, with patients presenting different autoimmune diseases. The effect was minor in patients with SSc, in whom the improvement was 4±47m, although the control group without treatment presented a greater deterioration of -37±120m. This benefit continued until two years of follow-up, according to the results from the PATENT-2 trial. Additionally, hemodynamic variables such as pulmonary vascular resistance improved, serum levels of NT-pro BNP were reduced, and NYHA functional class improved or became stable in 97% of patients, being 22% greater compared to the control group. There were no differences in two-year mortality compared with non-autoimmune PHT patients.
Sildenafil11 given at any dose improved the 6-minute walk distance test in 45-50 m (p = 0.001), reduced pulmonary pressures in 2-4mmHg according to the given dose (p>0.05), and improved the NYHA functional class in one level in 20-30% of patients, compared to placebo, according to the given dose (95% CI = 16-42%, p>0.05). Adverse events such as flushing, diarrhea and other gastrointestinal symptoms were increased.
Treprostinil12 improved hemodynamic variables such as cardiac index and pulmonary resistance (p = 0.006), improved the mean 6-minute walk distance to 25 m (p = 0.05) with a dose-dependent effect, being those patients who received more than 9ng/kg/min the ones with greater improvements. Dyspnea scores improved compared to placebo after 12 weeks of treatment. More side effects such as injection site reactions and those related with the use of prostanoids were identified; however, patients had an adequate tolerance in most cases, without the suspension of the treatment.
The use of Epoprostenol in the non-randomized clinical trial13 increased the mean 6-minute walk distance in 46 m, compared to a mean reduction of -48 m in the control group using conventional therapy, after 12 weeks of treatment. The 108 meters' difference was significant (95% CI = 58-180m, p = 0.001). They found a reduction in pulmonary pressures of 6 mmHg, and also in pulmonary vascular resistances. Twenty-one patients from the treatment group presented improvement in NYHA functional class, while no patients improved in the control group. Raynaud phenomenon and digital ulcers also improved in the patients receiving Epoprostenol. The case-control study10 does not allow making additional conclusions due to a limited sample of 4 patients, although it refers an improvement in mortality.
Combined therapy was assessed with patients from the AMBITION trial,8 in which the combined therapy of Ambrisentan plus Tadalafil was compared against monotherapy with the same agents. The primary outcomes including therapeutic failure (defined as a worsening in functional class, in 6-minute walk test, use of prostanoids, transplant or atrial septostomy requirement), occurred in less proportion in those patients receiving combined therapy (20%), compared to those receiving monotherapy (41%) (HR: 0.44; 95% CI = 0.22-0.89). Considering secondary outcomes, a reduction in NT-pro BNP levels and an improvement of 404±12 m in the 6-minute walk test were found. Regarding adverse events, peripheral edema was developed in 44% with combined therapy, compared to 26% of patients receiving Ambrisentan monotherapy and 33% of patients with Tadalafil monotherapy. No differences were found for the development of headache, anemia or syncope.
Most studies that included Bosentan were cohorts, followed by 3 controlled clinical trials (2 evaluated Bosentan vs. placebo, and 1 also evaluated Sitaxsentan), and one case-control study. An overall of 504 patients with SSc-PHT receiving treatment, and 148 patients without treatment for the control groups, were included.
The pilot clinical trial26 grouped 32 SSc patients with class III NYHA functionality, randomized in a 2:1 relation to Bosentan 125 mg every12 h or placebo. After 12 weeks of treatment, an improvement of 76 m in the 6-minute walk test (95% CI = 12-139 m; p = 0.02) was found. The improvement was maintained after 20 weeks of treatment. They also found an improvement in hemodynamic parameters including cardiac index and a reduction of vascular resistances (p = 0.002), and in preset outcomes (Borg dyspnea score, NYHA functional class, and medication withdrawal due to clinical worsening). However, this data was derived from the whole group of patients, and no stratification analysis was made to identify the improvements only for SSc patients.
The BREATHE-1 trial25 compared Bosentan vs. placebo in patients with PHT, including those with SSc. An improvement in the 6-minute walk test (44m; 95% CI = 21-67 m; p>0.001), Borg dyspnea score and WHO functional class was found.
Based on the subgroup analysis, the authors considered that Bosentan may have a preventive effect on patients with SSc, considering a decline in exercise capacity, with an improvement of 3 m compared to placebo (reduction of 40 m) using the 6-minute walk test. No significant differences were found for mortality. The safety profile showed that the presence of abnormal hepatic biochemical profiles was dose-dependent.
The 3-year follow-ups of the patients included in these trials were compared with historical controls, showing a one-year survival of 85%, and a two-year survival rate of 70% with Bosentan monotherapy. Patient's survival with Bosentan vs. Epoprostenol was 82% during the first year of treatment, and 67% during the second year, in 30% of patients. The follow-ups also showed that survival was lower in patients with SSc, compared to those with idiopathic PHT.21
The study that assessed the use of Sitaxsentan vs. placebo, and vs. Bosentan22 in the 6-minute walk distance used an open arm without blinding for Bosentan. Patients with PHT of different etiologies were included (30% of patients with connective-tissue diseases-associated PHT without specifying how many patients had SSc). For all patients, an improvement of 31.4m with a dose of 100 mg (p = 0.03); 24.2 m with a dose of 50 mg (p = 0.07), and 29.5 m with Bosentan (p = 0.05), was observed compared to placebo. Additionally, Sitaxentan improved the WHO functional class (p = 0.04).
Between the non-randomized studies, a retrospective cohort from the RESCLE Spanish registry was found.14 Of 1817 patients with SSc, 76 had SSc-PHT, receiving monotherapy with phosphodiesterase inhibitors or endothelin receptor antagonists as initial combined therapy, or as a combined sequential therapy. They compared mortality rates at 1, 3, 5 years between the groups. The sequential combination group had the lowest mortality (HR: 0.11; 95% CI = 0.03-0.5; p = 0.004). One-year survival was 78% for monotherapy, 94% for initial combined therapy, and 95% for sequential therapy; three-year survival was 40% for monotherapy, 51% for initial combined therapy, and 81% for sequential therapy; five-year survival was 31% for monotherapy, 34% for initial combined therapy, and 56% for sequential therapy (p = 0.007). No significant differences were found between the groups regarding the development of adverse events.
The study conducted by Lunay and colleagues,17 also analyzed mortality with the use of combined therapy with phosphodiesterase inhibitors. Survival data was compared with a cohort from the same study group that was previously published. One-, three- and five-year survival rates were 92%, 89% and 79%, respectively with Bosentan; and 80%, 56% and 51% respectively for patients with SSc-PHT (p = 0.001). Another study with mortality analysis23 shows differences between the control group and those patients using Bosentan. However, the control group was using prostacyclins, and patients had greater lung fibrosis, and worse functional capacity, making the results not reliable, as these conditions were not considered during the analysis.
The rest of case-control studies and cohort studies, reunited 449 patients, and they showed variable assessed outcomes. Two studies in patients with SSc-PHT assessed as main outcome the levels of NT-pro BNP,17,18 without finding a significant reduction after the first or second year using Bosentan. It was not related to the significant change in the NYHA functional class (p = 0.01), and the improvement in the 6-minute walk distance (p = 0.04). The other study with less patients found a reduction of NT-pro BNP after 7 months of treatment, but it was not statistically significant (p = 0.6).
A third study20 assessed the levels of NT-pro BNP in patients with SSc who developed digital ulcers and were treated with Bosentan. They also assessed the development of PHT using echocardiographic estimation, finding a reduction of the mean pulmonary pressure. No patients developed PHT in the group receiving Bosentan, compared to those from the control group in which 7 patients developed the condition. Patients from the Bosentan group achieved a reduction of pulmonary pressure values, while patients from the control group presented an increase of these values (p = 0.001). Borg dyspnea scores improved in the Bosentan group (p = 0.001) and the NT-pro BNP values decreased compared to the initial ones, in contrast to the control group (p = 0.004).
A cohort of 98 patients16 from the PHAROS registry database compared the effect of adding phosphodiesterase inhibitors to endothelin receptor antagonist monotherapy with Bosentan or Ambrisentan. The outcome of time to clinical worsening was defined as an increase in symptoms, death, hospitalizations, or need of prostanoids. Clinical worsening developed earlier in the group of patients with monotherapy, compared to those with combined therapy or phosphodi-esterase inhibitors. The multivariate analysis showed that the initial use of endothelin receptor antagonist monotherapy was associated with a shorter time to clinical worsening, compared to combined therapy or phosphodiesterase inhibitors (HR: 2.63; 95% CI= 1.28-5.56; p = 0.009). There were no significant differences between the groups at the beginning of follow-up.
Another study analyzed the combination of endothelin receptor antagonists and phosphodiesterase inhibitors in patients with PHT in whom Bosentan monotherapy failed. They found that in patients with SSc, the improvement in NYHA functional class and 6-minute walking distance was inferior. This same comparison between groups (SSc-PHT vs. idiopathic PHT) using Bosentan monotherapy24 found the same results, with better response outcomes in patients with idiopathic PHT vs. those with SSc, regarding the WHO functional class.
A development of a meta-analysis was attempted by the researchers, especially for those studies using Bosentan. However, the lack of specific data and the heterogeneity in the comparison groups did not allow it for outcomes such as dyspnea scores or NYHA functional class.
Two clinical trials of Bosentan vs. placebo assessed as main outcome the 6-minute walk test. In the study by Rubin and colleagues,25 data could be inexactly extracted as a graphic. However, in the study by Channick and colleagues26 no specific data for patients with SSc was given, making impossible the development of a meta-analysis. In the observational studies, a meta-analysis was not made regarding the 6-minute walk distance because: (1) one study did not use a control group as comparison15; (2) another study compared the results with a group of patients with idiopathic PHT19(3) compared the results with a control group receiving prostanoids.23
Discussion
After the systematic literature review, it was found that there is evidence available, derived from trials for the treatment of PHT that include patients with SSc. They show a beneficial effect with the use of different pharmacological agents, especially represented by the improvement in the 6-minute walk distance which was the most frequent test used during the follow-up of patients. The positive effects in the 6-minute walk test were seen with the use of endothelin receptor antagonists such as Ambrisentan and Bosentan (the last one had the greatest amount of studies using it), prostanoids, phosphodiesterase inhibitors such as Sildenafil, and Riociguat which also presents improvements in other outcomes including NYHA functional class and Borg dyspnea score. The outcome of mortality was also satisfactory in several studies, as survival rates presented an improvement compared to historical cohorts and even similar rates than patients with different etiologies of PHT with better prognosis such as those with idiopathic PHT. Likewise, several hemodynamic measurements and biochemical markers such as NT-pro BNP showed improvements during follow-ups.
Despite these findings, there are still several difficulties and interrogations to solve about this topic. More data is required to solve the following situations: identifying the best therapy for this group of patients, if these patients benefit more with an early combined therapy, if any of the pharmacological agents is superior to others, and the impact of non-pharmacological treatments such as rehabilitation. There is also limited data regarding patients with concomitant pulmonary pathologies which are frequent in those with SSc.
The benefits presented by the evidence available should be assessed with tools that could demonstrate also direct improvements over symptoms and quality of life, especially considering that the most frequent outcome assessed in the studies was the 6-minute walk test, which has some relationship with mortality in the initial measurements, but its variation from the initial measurement has limited prognostic significance. Even so, other hemodynamic and biochemical outcomes have greater prognostic value for mortality, but they were not frequently used in the studies included.
The overall quality of evidence for the studies included was moderate for clinical trials, according to GRADE classification, and in some cases it was high,7,11,12,16 as they were derived from the main trials of pharmacological treatment. The GRADE classification for the non-randomized trials was low, and in some cases very low given their risk of bias.
Regarding the development of a meta-analysis of selected studies, it was not possible in any case due to the design heterogeneity, the number of different pharmacological agents studied, the comparison populations, the differences in the follow-up times for the measurement of outcomes, and the absence of specific data for patients with SSc.
Conclusion
Evidence available regarding pharmacological treatment of SSc-PHT show that the use of medications approved is associated with an improvement in clinical and not clinical outcomes, favoring their implementation in the therapeutic schemes for this disease. Most studies suggest the use of combined therapy with multiple pharmacological agents to achieve a better response. However, the evidence available is still limited for patients with SSc, and future research is required to develop specific recommendations regarding the best pharmacological treatment and the best combination therapy.

