











 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkIntroduction
Vasculitides belong to a heterogeneous group of pathologies, whose main finding is the involvement of the walls of blood vessels of different sizes, which is the cause of damage to different organs. (1 Their presentation in the pediatric age is infrequent. In general, the data related to the clinic, classification and management are extrapolated from the adult population.
The consensus of the European League against Rheumatism/Pediatric Rheumatology European Society (EULAR/PReS) classifies the vasculitides in children according to the clinical phenotype (systemic or single organ involvement), the size of the affected vessel (small, medium or large) and the histopathology of the infiltrates. (2
Small-vessel vasculitides mainly affect capillaries and venules, although they can compromise arteries and veins of larger caliber. (3,4 They are divided into two entities: a) granulomatous vasculitides, which include pathologies such as granulomatous disease with polyangiitis (GPA) (formerly known as Wegener's granulomatosis) or Churg-Strauss syndrome (known today as eosinophilic granulomatosis with polyangiitis [EGPA]) and; b) non-granulomatous, which include microscopic polyangiitis (MPA), different from the classification in adults, which is given according to the Chapel-Hill Consensus Conference and divides small-vessel vasculitides into vasculitides associated with antineutrophil cytoplasmic antibodies (ANCA): GPA, MPA and EGPA, and vasculitides due to immune complexes, whose main representative is Henoch-Schönlein purpura (HSP).
The ANCAs described are the antibodies against the target antigen proteinase 3 (PR3-ANCA) and the myeloperoxidase antibodies (MPO-ANCA). (5 For epidemiological purposes, the Chapel-Hill classification has been useful and has been applied to the pediatric population. The determination of the ANCA report of patients with ANCA-associated vasculitis is imperative in clinical practice. (6
In pediatrics, ANCA-associated vasculitides (AAVs) have a heterogeneous clinical presentation, with frequent involvement of the respiratory tract, the kidney, the skin, and the joints. They are infrequent in childhood; from 0.5 cases/1,000,000 to 6.39 cases/1,000,000 have been reported, with a peak incidence in adolescence. The median age at diagnosis is approximately 11-14 years and a predominance in women has been observed (unlike the adult population in which AAVs are more common in men). (3,4
Within the small-vessel vasculitis, HSP is the most common in the pediatric age group with an incidence of approximately 20/100,000 children in developed countries and a peak incidence between four and six years. (5,6 The mortality rate due to AAV in childhood is 5-10%; patients with EGPA appear to have a higher estimated mortality rate, which ranges from 15% to 18%. Diverse studies have reported a rate of end-stage kidney disease of 29-40% in MPA and of 10% in GPA. (5 Taking into account the possibility of progression to end-stage kidney disease in these patients, with requirements for renal replacement therapy (RRT) and associated high mortality, the characterization of this entity in our population of children is essential.
AAV symptoms include constitutional symptoms in 50% of the patients (fever, fatigue, weight loss, and anorexia). Renal involvement is common and can range between hematuria, proteinuria, or rapidly progressive glomerulonephritis, which is considered a predictor of mortality, especially in those patients who debut with glomerular filtration rates (GFR) below 50mL/min/1.73m2, in whom it has been demonstrated a risk of up to 50% for kidney failure or death at five years of follow-up. (7
Due to the fact that the initial symptomatology in many cases is nonspecific, the diagnosis can be significantly delayed and, therefore, the delay in the establishment of medical management can influence the renal prognosis. (5
We present three cases of female patients under follow-up by the pediatric nephrology service, whose cases were known during hospitalizations in a fourth level institution in southwestern Colombia. AAV was confirmed in all of them by histopathological findings in biopsies and renal involvement; only one of them presented positive ANCA.
Case reports
First case: ANCA negative MPA
Patient who in her first years of life presented repeated episodes of urinary tract infection, without extension studies. Later, at the age of five years, she debuted with a clinical picture of edema, proteinuria and high blood pressure (HBP), for which she was hospitalized with a diagnosis of nephrotic syndrome (NS) in another institution. Paraclinical tests during hospitalization revealed hematuria, proteinuria and elevated levels of nitrogen compounds. At eight weeks, corticoresistant NS was considered due to the lack of response to steroid management and a renal biopsy was performed, which reported extracapillary proliferative glomerulonephritis with normal C3 and C4 serum complement.
Despite the management with cyclophosphamide pulses, the patient presented marked clinical deterioration which required peritoneal dialysis. A second biopsy was performed in our institution, with evidence of pauciimmune sclerosing glomerulonephritis with less than 10% fibrocellular crescents, interstitial fibrosis, and severe tubular atrophy (Figs. 1 and 2). Subsequently, the levels of MPO-ANCA and PR3-ANCA were measured by indirect immunofluorescence, which were negative.
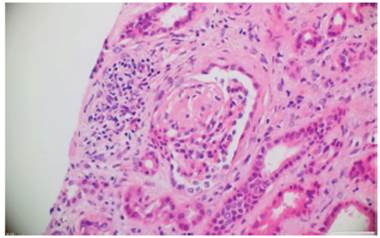
Fig. 1 Hematoxylin and eosin. Glomerulus that presents focal and segmental glomerulosclerosis. In the interstitium, edema and mononuclear inflammatory infiltrate with tubular atrophy are observed. 10x.
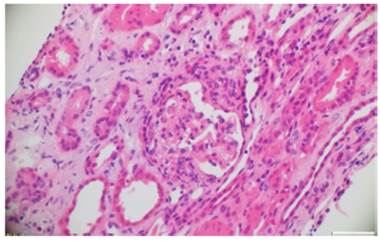
Fig. 2 Hematoxylin and eosin. Glomerulus with extracapillary proliferation and formation of fibroepithelial crescents that obstruct the glomerular tuft. 10x.
Four months after the second biopsy, and after confirmation of the diagnosis of ANCA-negative vasculitis, the patient presented with fever associated with influenza B infection and required mechanical ventilation. In addition, she presented severe anemization, pleural effusion, ascites and alveolar hemorrhage. The report of ANCA was negative: PR3-ANCA = 1 U/mL (negative <5U/mL) and MPO-ANCA 0.9U/mL (negative <5U/mL). The ANAs were also negative by immunofluorescence, while the report of anti-DNA antibodies (Crithdia luciliae substrate) was positive with one in 10 dilutions. The patient was evaluated by pediatric rheumatology, which considered an ANCA-negative vasculitis with diffuse alveolar hemorrhage (kidney-lung syndrome), for which she was managed with steroid pulses, cyclophosphamide (750mg/m2), plasmapheresis, acute hemodialysis and subsequent peritoneal dialysis. She had clinical stabilization and continued outpatient management with azathioprine (3 mg/kg/day), prednisolone (1 mg/kg/day) and enalapril (0.25 mg/kg/day) for nine months.
13 months after the diagnosis of vasculitis, the patient underwent kidney transplantation from a deceased donor and the clinical evolution was satisfactory. She is currently 11 years old and attends intra-institutional monthly follow-up with a glomerular filtration rate of 80mL/min/1.73m2, and receives tacrolimus (dose of 0.15 mg/kg/day) and mycophenolate mofetil (dose of 600 mg/m2/day) as immunosuppression.
Second case: ANCA positive MPA
A 13-year-old patient with a clinical picture of two months of evolution of progressive mucocutaneous pallor associated with a febrile syndrome of 20 days of evolution and asthenia. The physical examination revealed the presence of suppurative nodules on the scalp and pharyngeal erythema. She also presented arthralgia in the left knee with vasculitic lesions, violaceous papular lesions that did not disappear with digito-pressure.
The paraclinical tests on admission showed anemia (hemoglobin of 8.9 g/dL), hematuria and proteinuria (75 mg/dL) in a spontaneous urine sample, elevation of nitrogen compounds (BUN = 45 mg/dL, serum creatinine = 2.9 mg/dL), normal serum complement C3 and C4, negative antinuclear antibodies and antiglomerular basement membrane antibodies, negative PR3-ANCA <3.5 U/mL (negative <5 U/mL) and positive MPO-ANCA 72U/mL (negative <5U/mL). On admission, complicated post-streptococcal glomerulonephritis versus lupus nephritis were suspected. Given the progressive increase in nitrogen compounds and the clinical deterioration of renal function, it was required to start hemodialysis and perform a renal biopsy.
The renal pathology showed active crescentic proliferation of cellular origin, with high activity, extensive fibrinoid necrosis, leukocyte infiltration, and evidence of lymphoplasmacytic infiltrate in the interstitium, associated with fibrosis in 10%. There was no evidence of deposits of immune complexes (Figs. 3 and 4).
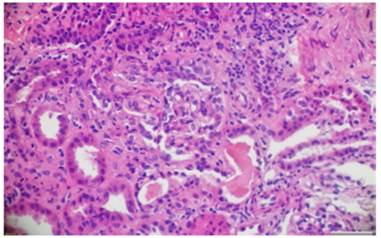
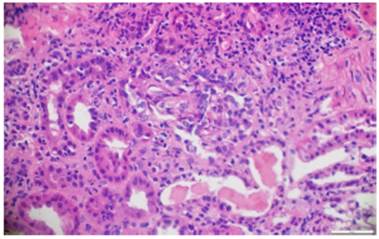
Fig. 4 Hematoxylin and eosin. Fibrocellular crescents with obliteration of the capillary lumen in the glomerular tuft.10x.
The patient continued with impaired renal function. Along with pediatric rheumatology and pediatric nephrology, it was decided to start management with plasmapheresis, cyclophosphamide, and rituximab. She received a total of six doses of cyclophosphamide at 10mg/kg/dose: the first three doses every two weeks and subsequently the other two doses every three weeks; and four doses of rituximab: a weekly dose of 375 mg/m2/dose. Azathioprine was discontinued due to leukopenia and mycophenolate mofetil (MMF) was continued (1100 mg/m2/day). The patient presented an adequate clinical evolution and improvement in renal function, for which hemodialysis was discontinued three months after starting immunosuppressive management.
The patient is currently 18 years old, is under follow-up by rheumatology with a diagnosis of MPA, remains asymptomatic, has a serum creatinine of 1 mg/dL and is managed with MMF (1100mg/m2/day).
Third case: ANCA negative GPA
The third case occurred in a seven-year-old patient who debuted with a clinical picture of two years of evolution consisting of repeated episodes of respiratory infection and stridor, managed on an outpatient basis with steroids on multiple occasions. Three months before admission, the patient presented hyporexia, weakness, and macroscopic hematuria. Subsequently she developed generalized edema. On physical examination, she presented a left intraorbital mass, originating from the lacrimal gland and the lesions described above.
During the evolution, she presented HBP, nephrotic-range proteinuria, normal C3 and C4, and negative antinuclear and anti-DNA antibodies. ANCA test was performed by IIF, which reported positive PR3-ANCA (1:160 dilutions) and negative MPO-ANCA. However, MPO-ANCA and PR3-ANCA were carried out by ELISA, which were negative (PR3-ANCA <0.2 U/mL [negative <5 U/mL] and MPO-ANCA 0.8 U/mL [negative <5 U/mL]).
It was considered that the patient had GPA and immuno-suppression was started with cyclophosphamide (a total of six doses of 10 mg/kg/dose, the first three doses every two weeks and thereafter the other two doses every three weeks) and rituximab (four weekly doses of 375mg/m2/dose). However, despite the established management, the patient presented a rapid deterioration of renal function and required peritoneal dialysis.
At the age of 10 years, she presented multiple complications derived from granulomas in the skin, mucous membranes (including the esophagus), soft tissues, and bones.
The skin biopsy of the ulcerative lesion on the left thigh had a pathology report that showed abundant mixed inflammatory infiltrate with suppurative polymorphonuclear predominance, areas of extensive ulceration, granulation tissue with proliferation of vessels, and abundant histiocytes. In addition, the renal biopsy reported the presence of fibrocellular crescents in more than 50% of the glomeruli, proliferation of mesangial cells, and immunofluorescence negative for immune complexes (Figs. 5 and 6).
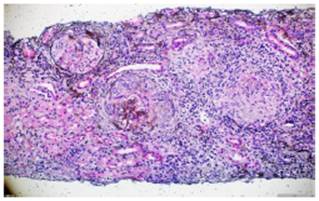
Fig. 5 Silver methenamine. Glomeruli with presence of fibrocellular crescents and fibrinoid necrosis in the glomerular tuft. 10x.
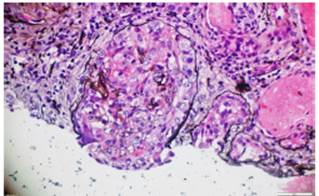
Fig. 6 Silver methenamine. Fibrous crescent with destruction of the glomerular basement membrane and replacement of epithelial cells by fibroblasts in a scarring process. 40x.
The patient presented an important generalized deterioration, with sclerosing peritonitis as a major complication, which forced a change of RRT from peritoneal dialysis to hemodialysis. She died in the context of sepsis due to invasive fungal disease.
Discussion
The term vasculitis, as already mentioned, is used to identify a heterogeneous group of pathologies whose main finding is the involvement of the walls of the blood vessels. (1
Specifically for the diagnosis of Wegener's granulomatosis, the EULAR/PReS consensus described the need for positivity of at least three criteria (alterations in the urinalysis such as significant hematuria/proteinuria, presence of granulomatous inflammation in the biopsy, inflammation of the nasal sinus, subglottic, tracheal or endobronchial stenosis, abnormal chest x-ray or tomography and ANCA positive staining).2
In contrast, vasculitides in adults are classified according to the Chapel-Hill consensus, in which in turn they are grouped according to the size of the involved vessel, into large, medium and small vessels, variable vessel vasculitis, and single-organ vasculitis, associated with systemic disease or associated with probable etiology.
For small-vessel vasculitides, the terms granulomatosis with polyangiitis (Wegener's granulomatosis), eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome) and microscopic polyangiitis appear, all under the same denomination of AAV. (8 ANCA-negative AAVs are a subgroup with clinical and histological characteristics of AAV, but with negative antibody reports; the literature describes an apparent less severe systemic or limited renal involvement in these patients. (7
Renal involvement in the context of AAV can manifest as hematuria, proteinuria, hypertension, or rapidly progressive glomerulonephritis. GPA is characterized by granulomatous inflammation and necrosis. Although it has a predilection for the respiratory tract (80%), the kidney can also be severely compromised in up to 75% of cases. (2,9 It is associated with MPO-ANCA in 20%, PR3-ANCA in 75%, and ANCA negative in 5%.7,10
The third patient also had a debut with hematuria, added to proteinuria and hypertension, a report of negative MPO-ANCA and PR3-ANCA by ELISA, renal biopsy with a predominance of crescents, and therapeutic failure requiring peritoneal dialysis. In Colombia, there is a pediatric case of clinical debut described with alveolar hemorrhage, hematuria, oliguria, requirement for renal replacement therapy and high PR3-ANCA titers, added to renal biopsy findings of compromise by focal and segmental necrotizing glomerulonephritis with extracellular proliferation and presence of crescents in more than 70% of the glomeruli, without recovery of renal function, despite management with steroids, cyclophosphamide and plasmapheresis. (11 Other Colombian series report management with rituximab in pediatric patients with GPA without severe renal involvement. (12
With regard to the report of ANCA in the third case by positive IIF and by negative ELISA, the literature describes that the techniques to measure ANCA positivity are ELISA and immunofluorescence (IIF). If both tests show positivity for ANCA, the positive predictive value (PPV) is described in up to 88% of the cases. The IIF test has a degree of subjectivity, since it is based on visual assessment of the immunofluorescence pattern, so the result may depend on the expertise of the reader. Regarding the ELISA technique, it is associated with higher percentages of specificity (E) and PPV, compared to immunofluorescence (E 82% and PPV 63% vs. E 18% and PPV 13%, respectively). (13,14 In the adult population IIF and ELISA are generally used in combination. (15
MPA is less frequent in children and, unlike GPA, does not present granulomatous inflammation. (9 It is associated with MPO-ANCA in 60%, with PR3-ANCA in 30%, and with negative ANCA in 10% of the cases. (7 In pediatrics, skin involvement of purpura type is a common presentation, and neurological (21-86%) and pulmonary (44%) involvement are also common. Renal involvement (75-90%) can be rapid and severe, with the possibility of requiring RRT in more than half of the patients. Up to 25% of patients may have HBP and mortality at one year is up to 85%.16,17
The patients in the first and second cases debuted differently from the renal point of view: the first one as negative NS, MPO-ANCA and PR3-ANCA and a history of management with steroids, cyclophosphamide and plasmapheresis; and the second, with non-severe hematuria and proteinuria, positive MPO-ANCA and a history of management with steroids, cyclophosphamide, plasmapheresis and rituximab. However, both suffered deterioration that required RRT, one of them without recovery and requiring kidney transplantation and the other with recovery of kidney function three months after the start of the hemodialysis.
EGPA, a disease that generates granulomatous inflammation rich in eosinophils, is even less common in children and has been associated with asthma and atopy. (1 Renal involvement is usually mild and is associated with MPO-ANCA in 45%, PR3-ANCA in 5%, and ANCA negative in 50%.7,18 It is the vasculitis that presents the highest proportion of negative ANCA if there is no renal involvement, and in our pediatric nephrology service we have not had representative cases.
The diagnosis of the renal commitment is established by biopsy. The AAVs cause glomerulonephritis that is characterized by infiltrates in the vessel walls, with predominance of polymorphonuclear leukocytes (neutrophils, eosinophils). (1 The histopathological findings have a good clinical correlation, the distinctive histopathological pattern is the presence of necrosis in the capillary loops, extracapillary proliferation with formation of crescents or half-moons, interstitial and periglomerular infiltrate, necrotizing arteritis and absence or pauciimmunity of immune complexes. (1,19
Berden et al. devised a histopathological classification system to confirm ANCA-associated glomerulonephritis, called focal (more than 50% of normal glomeruli), crescentic (more than 50% of glomeruli with crescents), mixed (less than 50% of normal glomeruli, less than 50% with crescents and more than 50% with global sclerosis) and sclerosing (more than 50% of glomeruli globally sclerosed). (20 For patients classified as focal, renal survival at one year is 93%, 84% for patients classified as crescentic, 69% for patients classified as mixed, and 50% for those classified as sclerosing.
The cases reported here had variability in the histological report: the one classified as GPA had crescents in more than 50% of the glomeruli, in concordance with loss of renal survival and the need for dialysis. Those classified as MPA presented different histological findings. The ANCA negative MPA had as an outcome impaired renal function and kidney transplantation. In these cases, interstitial fibrosis and severe tubular atrophy predominated, while in the positive ANCA MPA, fibrinoid necrosis prevailed, with recovery of renal function after the renal relapse.
In general, the renal prognosis is related to multiple findings, some of which are histologically predominant, such as the number of normal glomeruli, the degree of tubulitis and interstitial inflammation, and the presence of sclerosis or crescents/half-moons in the renal biopsy. Follow-up studies describe the possibility of developing end-stage kidney disease in up to 40% of patients with MPA and in 10% of those with GPA.5
Conclusions
AAVs are a group of rare multisystemic diseases in the pediatric age, whose general symptoms at the beginning make diagnosis difficult and nonspecific in early stages. The clinical variability and the different renal outcomes of the same pathology stand out in the three cases presented, that include recovery of renal function, renal replacement therapy, transplantation and death.

