











 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkINTRODUCTION
Equine herpesvirus 1 (EHV-1) and 4 (EHV-4) are important pathogens that have a significant economic impact on horse populations worldwide. They are responsible for a variety of diseases, including respiratory disease, abortion, neonatal disease and myeloencephalitis 1. The two viruses have seroprevalence rates ranging from 9 to 28% for EHV-1 and 90 to 100% for EHV-4 1,2.
Both viruses are members of the family Herpesviridae, subfamily Alphaherpesvirinae and genus Varicellovirus2. The viruses have double stranded DNA genomes containing 150,223 and 145,597 base pairs (bp) in length, respectively 1,3, and 79 and 80 open reading frames (ORFs), respectively 1. Infection by these alphaherpesviruses produces a respiratory disease characterized by fever, anorexia and nasal and ocular discharge, which, together with bacterial proliferation, may contribute to the production of rhinopneumonitis 1,2. Specifically, EHV-1 replication occurs in epithelial cells of the upper respiratory tract and in local lymph nodes, resulting in leukocyte-associated viraemia 2. Such viraemia leads to viral replication in the endothelial cells of blood vessels and in the central nervous system and pregnant uterus, thus triggering abortions and paresis 2,4. It has been proposed that EHV-1 has neuropathogenic and non-neuropathogenic strains differentiated by a single mutation in ORF30 encoding viral DNA polymerase 2,5.
Although the pathogenesis of EHV-4 is still unclear, it is a cause of respiratory disease in horses 1,2, leukocyte-associated viremia is rare and occasionally associated with abortion and neurological disorders 4,6. One of the main characteristics of alphaherpesviruses is the development of latency in lymphoid tissues, circulating lymphocytes and trigeminal ganglia (TG) 2,7. Latent infection implies that the viral genome is in a non-replicative, restricted transcription state wherein only Latency-Associated Transcripts (LATs) are produced 8. EHV-1 and 4 are in episomal form in TG, or in lymphoid cells 9,10. Both for EHV-1 and for EHV-4, specific LATs from genes 64 and 63 have been found in peripheral blood mononuclear cells (PBMC) and trigeminal ganglia, respectively 10.
EHV-1 and EHV-4 can be diagnosed directly through the detection of virus in clinical samples (nasal swabs, serum and buffy coat samples) by virus isolation in cell culture, conventional or real time PCR 11,12 or indirectly through the detection of EHV-1 or EHV-4 antibodies in serum or cerebrum spinal fluid (CSF) by viral neutralization, complement fixing or EHV-1/EHV-4 type specific ELISA based on recombinant antigens of glycoprotein G (gG), thus allowing differentiation between them 11,12.
However, these tests only indicate whether the horse has been exposed to the virus 12,13. PCR assays are more sensitive and rapid, have replaced the time-consuming procedure of virus isolation 13,14, however these tests are unable to differentiate between a replicating or non-replicating virus 14,15. For this reason, diagnosis of latently infected horses is important because they represent virus reservoirs 16, as these tests are based on molecular detection of LATs and quantification by real time PCR at a DNA and mRNA level, in order to differentiate viral states 10,14.
In Colombia, the first report of EHV-1 was in 1992, when the virus was isolated from samples of an aborted fetus whose dam had come from Argentina 17. In 2007 and 2008, 18,19 a seroprevalence was reported in the regions of Antioquia and Meta, with co-infection with both viruses. The objective of this study was to evaluate the presence of EHV-1 and EHV-4 in a selected population of Colombian horses, through serology, detection of viral genomes in submandibular lymph node and trigeminal ganglion samples and detection of LATs in both tissues.
MATERIALS AND METHODS
Animals and sample collection. The serum and tissue samples were taken from horses (n = 50) slaughtered in a commercial slaughterhouse. The equine abattoir is located in the department of Cundinamarca in the center of Colombia, where it receives horses from all over the country. Of the fifty horses, 24 were males and 26 were females, with an age distribution that varied between 5 to 25 years old estimated by photos of incisor dentition. Blood samples (10-15ml) from each slaughtered horse were taken and transported to the laboratory, where the samples were centrifuged at 2500 rpm for 10 min and sera were stored at -70˚C. Submandibular lymph nodes (SLN) and trigeminal ganglia (TG) were recovered by horse head dissection, transported at 4˚C to the laboratory and stored at -70˚C. To avoid cross-contamination among tissues from different horses, disposable instruments were employed to take each sample. The status of vaccination of the horses studied for EHV-1 and 4 was unknown.
Antibodies detection for EHV-1 and EHV-4. Serum samples were tested for the presence of antibodies to EHV-1 and EHV-4 using the SVANOVIR EHV-1/EHV-4 Ab kit (Svanovir, Sweden), according to manufacturer’s instructions. The results were recorded at 450nm using a (BioTek® Power Wave XS OD) ELISA reader. Samples were considered positive at >0.2 and negative at lower than 0.1.
Nested PCR (nPCR). Twenty-five milligrams of each tissue (TG and SLN) were excised and the DNA was extracted using the QIAamp® DNA mini kit (QIAGEN®), in accordance to the manufacturer´s instructions. The nPCR amplified a conserved region of the gB gene for both EHV-1 and EHV-4, primers reported by Borchers et al, 1993 (20). EHV-1 primers for the first PCR reaction were (P1 5´-TCTACCCCTACGACTCCTTC-3´ and P2 5´-ACGCTGTCGATGTCGTAAAACCTGAGAG-3´) and for the second PCR reaction were (P3 5´-CTTTAGCGCTGATGTGGAAT-3´ and P4 5´-AAGTAGCGCTTCTGATTGAGG-3´), which amplified a region of 1474 and 771pb, respectively (20). EHV-4 primers for the first PCR reaction were (P1 5´-TCTATTGAGTTTGCTATGCT-3´and P2 5´-TCCTGGTTGTTATTGGGTAT-3’) and for the second PCR reaction were (P3 5´-TGTTTCCGCCACTCTTGACG-3´and P4 5´-ACTGCCTCTCCCACCTTACC-3´), which amplified a region of 952 and 600pb, respectively (20). Reactions were performed in a total volume of 25 µl containing 0.25 µl of Taq polymerase (5 U/µl) (Go taq flexi - Promega®), 5x Taq buffer (2.5 µl), 2 mM MgCl2, 0.5 mM dNTP, 1 µl of each primer (20 µM) and 2 µl of extracted DNA. Positive and negative controls were included. The PCR reactions were performed on a Biorad® - DNA thermocycler using a protocol consisting, in a first round, of denaturation at 94ºC for 5 min, followed by 40 cycles including a denaturation at 94ºC for 1 min, annealing at 57ºC or 60ºC for 1 min for EHV-1 and EHV-4, respectively, and extension at 72ºC for 1 min, with a final extension at 72ºC for 10 min. One microliter of the first amplification reaction was used for the nPCR. The second-round PCRs were performed as described above, with annealing steps at 56ºC or 55ºC for 1 min for EHV-1 and EHV-4, respectively. The positive controls used corresponded to a Colombian EHV-1 cell culture-isolate from 1992 (17) and an EHV-4 positive clinical sample. DEPC-treated water was used as a blank control.
LATs detection by nPCR. TG and SLN samples testing positive or negative for EHV-1 or/and EHV-4 DNA detection were subjected to gene-63 and gen-64 LAT amplification by another nested PCR, taking into account that the primers for genes 63 and 64 amplify sequences for EHV-4 and EHV-1, respectively 21,22. For this purpose, RNA was extracted from tissues samples with the RNeasy® kit (QIAGEN), in accordance with the manufacturer´s instructions. All RNA samples were treated with DNase to digest any contaminating viral DNA. Complementary DNA (cDNA) synthesis reaction was performed using random hexamer primers (Promega®) and M-MLV (Invitrogen®), according to the manufacturers’ instructions. For the gen 63 nested PCR, the first reaction was performed with primers (63eF-5´ GGGGCAAGGGCTCTAAACCT-3´and 63eR-5´ CAGGAGACACCAGCAACGAC-3´) and the second PCR reaction with primers (63iF 5´-CAAACTCCCGCAGGTTGTATC-3’ and 63iR 5’-ACTTTGGACAGCGAGGGTGAA-3’), in order to amplify a sequence of 532 and 253bp, respectively (10). For the Gen 64 nPCR, the primers for the first PCR were (64eF 5´-GGAGACCGCGTCCAGCACTA-3´and 64eR 5´-CTCCGAGGGAAGCCAGACCT-3’) and for the second PCR reaction were (64iF 5´-GGACCCCCTGGGCGTTGAGG-3´and 64iR 5´-CCGCGGAGACTGCCACACTC-3´), which amplified a sequence of 1108 and 500 pb, respectively (10). The reactions were performed in a total volume of 25µl containing 0.25 µl of Taq polymerase (5 U/µl) (Go taq flexi - Promega®), 5x Taq buffer, 2 mM MgCl2, 0.5 mM dNTP, 1 µl of each primer (20 µM) and 2µl of cDNA. PCR reactions were performed on a Biorad® - DNA thermocycler. The first-round PCR consisted of an initial denaturation step at 95ºC for 5 min, followed by 40 cycles of denaturation at 94ºC for 30 seg, annealing at 55ºC and 64ºC for 45 seg for Gen 63 and Gen 64, respectively, and extension at 72ºC for 1min, with a final extension at 72ºC for 5 min. One microliter of the first amplification reaction was used for the nPCR. The second-round PCR was performed as described above, with the annealing step at 58ºC and 60ºC for 45 seg for Gen 63 and Gen 64, respectively. As positive controls, RK-13 cells were infected with EHV-1 (isolated in 1992 -Colombia) and at 48 hours post infection, the cells were harvested, pelleted, and then the RNA was extracted. For EHV-4, a positive clinical sample (by PCR) was employed. DEPC-treated water was used as a negative control for the DNA extraction and PCRs. Controls were included in each PCR run, and the controls from the primary PCR was carried over to the nested step.
Ethics committee. The current research was approved by the Ethics Committee of the Faculty of Veterinary Medicine and Animal Science of the Universidad Nacional de Colombia, Bogota. Equine slaughterhouse sacrifice protocols are under government legal authorization (Resolution 0222 of the Colombian Health Ministry).
Statistical analysis. Data were analyzed by Kruskal-Wallis. Probabilities p<0.05 were considered significant. All statistical analyses were performed using Statistics 8, software.
RESULTS
Serological reactivity. Serological assessment showed that 44/50 (88%) and 6/50 (12%) of horses were positive for EHV-4 and EHV-1 antibodies, respectively (Figure 1). It is noteworthy that all horses positive for EHV-1 antibodies were also positive for EHV-4. Among the seropositive horses, no difference was found between males and females for both viruses. Likewise, no age differences were observed in the presence of antibodies against EHV-1 and EHV-4 (p>0.05).
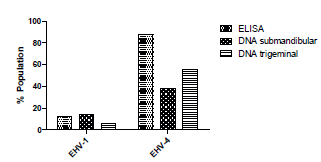
Figure 1 EHV-1 and EHV-4 serological reactivity by ELISA and viral DNA detection in submandibular and trigeminal tissues in the evaluated population
EHV-1 and EHV-4 in sub-mandibular lymph nodes and trigeminal ganglia. EHV-1 DNA was detected in 10/50 (20%) of horses (Table 1), and of the latter, horses were either positive in sub-mandibular lymph nodes (7/10) or in trigeminal ganglia (3/10). In the case of EHV-4 DNA, it was found in 30/50 of horses (60%), of which 17/30 (56%) were detected in both tissues (Table 1). EHV-1 and EHV-4 co-infection was detected in 5/50 horses (10%). Neither EHV-1 nor EHV-4 DNA detection revealed differences associated with age or sex.
Table 1 Detection of EHV-1 and EHV-4 DNA in trigeminal ganglia and submandibular lymph nodes
| Sample | EHV-1 DNA | EHV-4 DNA |
|---|---|---|
| Positive % (n/total) | ||
| Submandibular lymph nodes | 14 (7/50) | 38 (19/50) |
| Trigeminal ganglia | 6 (3/50) | 56 (28/50) |
| Trigeminal ganglia and submandibular lymph nodes | 0 (0/50) | 34 (17/50) |
| Total | 20 (10/50) | 60 (30/50) |
Comparatively, in the case of EHV-4, 44 (88%) horses were positive by ELISA and 30 (60%) by viral DNA amplification. In the evaluated population, the presence of this virus was determined in 38% (19/50) when evaluating the submandibular lymph nodes and 56% (28/50) in the trigeminal ganglion (Figure 1). In contrast, for EHV-1, 6 (12%) horses were sero-positive and 10 (20%) amplified viral DNA. Regarding detection of the virus in the mandibular nodule, it was found in 7/50 (14%) of the horses, whereas for the trigeminal ganglion it was 3/50 (6%). Among horses that tested negative by ELISA, 88% (44/50) for EHV-1 and 12% (6/50) for EHV-4), 5/44 (11.3%) were found positive for EHV-1 DNA amplification.
Analysis of the results for both ELISA and PCR for EHV-1 shows that the most frequent combination found was negative (38 horses). Likewise, EHV-1 positive results for all tests or positive for viral DNA in both tissues and negative for ELISA were never observed (Table 2). In contrast, for EHV-4 the most frequent combination was positive for all tests, together with negative for viral DNA detection and positive for ELISA.
Table 2 The results of EHV-1 and 4 testing to ELISA, PCR to EHV-1 and EHV-4 and LATS. Abbreviations denote horses that tested positive (+) or negative (-) for EHV-1/EHV-4 DNA or Gen 63/64 LATs. Positive ELISA in red.
| Horse ID | ELISA | EHV-1 PCR | EHV-4 PCR | LATS | ||||
|---|---|---|---|---|---|---|---|---|
| EHV-1 | EHV-4 | SLN | TG | SLN | TG | Gene 63 | Gene 64 | |
| 1 | 0.511 | 0.394 | + | - | - | + | - | + |
| 2 | 0.029 | 0.696 | - | - | + | + | + | - |
| 3 | 0.021 | 1.245 | - | - | + | + | + | - |
| 4 | 0.052 | 0.281 | - | - | + | + | + | - |
| 5 | 0.05 | 0.562 | - | - | + | + | + | - |
| 6 | 0.096 | 0.941 | - | - | + | + | + | - |
| 7 | 0.256 | 0.632 | + | - | + | + | + | + |
| 8 | 0.094 | 0.826 | - | + | + | + | - | - |
| 9 | 0.025 | 0.355 | - | - | + | - | - | - |
| 10 | 0.507 | 0.244 | + | - | - | - | - | + |
| 11 | 0.096 | 0.898 | - | - | - | - | - | - |
| 12 | 0.098 | 0.621 | - | - | - | - | - | - |
| 13 | 0.034 | 0.326 | - | - | - | + | - | - |
| 14 | -0.026 | 0.052 | - | - | - | - | - | - |
| 15 | -0.017 | 0.003 | + | - | - | - | - | + |
| 16 | 0.039 | 0.637 | - | - | + | + | + | - |
| 17 | 0.41 | 0.321 | - | - | - | + | - | - |
| 18 | 0.864 | 0.413 | - | - | - | - | - | - |
| 19 | 0.023 | 1.161 | - | - | - | - | - | - |
| 20 | 0.1 | 0.309 | - | - | + | + | - | - |
| 21 | 0.021 | 0.276 | - | - | + | + | - | - |
| 22 | -0.001 | 0.203 | - | - | + | + | + | - |
| 23 | -0.002 | 0.256 | - | + | - | - | - | - |
| 24 | 0.020 | -0.001 | - | - | - | - | - | - |
| 25 | 0.034 | 0.501 | - | - | + | + | + | - |
| 26 | 0.916 | 1.097 | - | + | - | + | - | - |
| 27 | 0.025 | 0.519 | - | - | - | + | + | - |
| 28 | 0.084 | 0.269 | - | - | + | + | + | - |
| 29 | 0.02 | 0.092 | - | - | - | - | - | - |
| 30 | -0.002 | 0.311 | - | - | - | + | + | - |
| 31 | -0.014 | 0.242 | - | - | - | - | - | - |
| 32 | 0.008 | 0.996 | - | - | - | - | - | - |
| 33 | 0.004 | 0.017 | - | - | - | - | - | - |
| 34 | 0.001 | 0.769 | - | - | - | - | - | - |
| 35 | 0.001 | 0.419 | - | - | + | - | - | - |
| 36 | 0.079 | 1.055 | + | - | - | - | - | + |
| 37 | 0.091 | 0.087 | - | - | - | - | - | - |
| 38 | 0.033 | 0.324 | + | - | - | - | - | - |
| 39 | 0.098 | 0.302 | - | - | - | - | - | - |
| 40 | -0.008 | 0.457 | - | - | + | + | + | - |
| 41 | 0.100 | 1.811 | - | - | + | + | - | - |
| 42 | 0.008 | 0.211 | + | - | - | + | + | - |
| 43 | 0.010 | 1.009 | - | - | - | - | - | - |
| 44 | 0.013 | 0.3 | - | - | - | + | - | - |
| 45 | 0.013 | 0.399 | - | - | - | + | + | - |
| 46 | 0.033 | 0.245 | - | - | + | + | - | - |
| 47 | 0.018 | 0.549 | - | - | - | + | + | - |
| 48 | 0.061 | 0.877 | - | - | + | + | - | - |
| 49 | 0.011 | 1.082 | - | - | - | + | + | - |
| 50 | 0.018 | 0.489 | - | - | - | - | - | - |
LATs detection. LATs of genes 63 and 64 were detected by RT-PCR in 22/50 (45%) of horses evaluated that horses were positive for virus detection by PCR. Gene-63 transcript was detected only from trigeminal ganglion in 17/28 (60%) of EHV-4 positive horses; whereas gene 64 transcript was amplified only from submandibular lymph node samples from 5/7 (71%) EHV-1 positive horses. However, transcripts for genes 63 and 64 were detected simultaneously only for one horse.
DISCUSSION
The aim of this research was to evaluate the occurrence of EHV-1 and EHV-4 in Colombia using serology and genome detection. Initially, our data confirmed that both viruses are present in the country, as previously reported 18,19, and in terms of prevalence it is in agreement with studies from other countries in the latest years 9, where the prevalence of EHV-1 was lower (12 to 21%) than EHV-4 (88 to 100%). The results of our study echoed previous reports 18,23 showing that virus distribution was unaffected by sex and age. In contrast to other studies 19, ours shows the presence of EHV-1 and EHV-4 viral genomes in sera-negative samples; this finding suggests that serological results do not indicate the stage of the disease because some horses with latent infection have failed to produce antibodies 24. Another possibility is that horses tested by ELISA might have been recently infected with EHV-1 and EHV-4 and have not yet mounted a detectable antibody response.
For the molecular detection of EHV-1 and EHV-4, the recommended samples are respiratory tract cells, serum, lymph nodes tissues and peripheral blood mononuclear cells (PBMC) 11,25, where the infectious virus is replicating. For the detection of latent virus, trigeminal ganglia and PBMC are the recommended tissues in order to detect markers of latency (LATs) 26,27, and28. In this study, EHV-1 and EHV-4 viral DNA were detected from submandibular lymph nodes and trigeminal ganglia, being that the percentage of viral genome detection for EHV-1 in submandibular lymph nodes was higher compared to TG, as reported by Dunowska et al, 2015 9. In the case of EHV-4, we found more viral DNA from TG, as was reported previously 18.
Consistent with previous studies 16,27, who report latency of 40 and 45% respectively; our study found LATs in 45% of infected horses. However, there are discrepancies between the studies regarding the site of latency for EHV-1 and EHV-4 9,27; specifically, EHV-1 has been reported to establish greater latency in lymphoid tissue than in nervous tissue. In the current study, EHV-1 LATs were only found in submandibular lymph nodes, whereas EHV-4 LATs were found in trigeminal ganglia. Several studies have shown that the technique used for detection may influence the ability to identify horses in latency, particularly using conventional PCR 9. Allen et al 2008 16, estimated the prevalence of EHV-1 latency using ultralow magnetic bead-based detection, sequence-capture, and nested PCR, finding about a 30% greater prevalence compared to conventional PCR. Based on this report, it is probable that in our study the number of horses in the latency phase is underestimated. It should be noted that herpesviruses produce latent infection, which reactivates under stress conditions 22,28. In the case of the population evaluated in this study, it should be noted that it was subjected to severe stress generated by long travel distances, extreme temperature changes, lack of food and overcrowding; these factors could activate viral replication 29. These particular study factors could indicate that the detection of LATs in TG and SNL cells demonstrate a latent form of infection coincident with the reactivation of active infection. For this reason, it is impossible to demonstrate the absence of an active EHV-1 and EHV-4 infection based solely on the detection of LATs, making it necessary to simultaneously include other diagnostic tests, such as the detection of mRNA encoding one of the viral glycoproteins, particularly if stress events are coincident with sampling.
With the techniques used in the present study, eight possible combinations of results were found (i.e., horses testing positive to genome detection in both studied tissues, but negative to ELISA or vice versa). These combinations clearly showed how a single diagnostic test is not sufficient enough to establish the status of a farm or even determine if the horse is under acute or latent infection. In addition, we found that horses seropositive for both herpesviruses also harbored the two viral genomes; this co-infection data is in agreement with previous reports 24,30. It is important to keep in mind that a seronegative horse is not indicative of viral absence, since it could have a latent infection. For this reason, it is important to add to serology the molecular detection of the viral genome and to demonstrate the presence or absence of LATs, which could help to establish a real status of the disease.
In conclusion there is still limited information concerning the presence and the prevalence of EHV-1 and EHV-4 in Colombia. However, the current study not only validated the endemic nature EHV-4, but also highlighted the circulation of EHV-1 with no evidence of prior immunization. Furthermore, although this study did not differentiate between active and latent EHV-1 and EHV-4 infection, the evidence of LATs and the presence on viral DNA in the same sample could be indicating a status of the disease.

