










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Médica de Risaralda
Print version ISSN 0122-0667
Revista médica Risaralda vol.20 no.1 Pereira Jan./June 2014
Reporte de Caso
Síndrome Carcinoide: Presentación de caso y revisión de la literatura
Jesús Hinestroza Barrios,1,3 Diego Alejandro Medina Morales,2* Milton Paredes Mendoza,3 Carlos Andrés Trejos Ramírez,3 Luis Fernando Valladales Restrepo.4
1 Clínica Saludcoop, Pereira, Risaralda, Colombia.
2 ESE Hospital San Rafael, Pueblo Rico, Risaralda, Colombia.
3 Facultad de Ciencias de la Salud, Universidad Tecnológica de Pereira, Pereira, Risaralda, Colombia.
4 ESE Hospital San Vicente de Paul, Santuario, Risaralda, Colombia.
* Correo electrónico: alejo1287_9@hotmail.com
Fecha de Recepción: 15-02-2013.
Fecha de Aceptación: 19-10-2013.
Resumen
Se presenta el caso de una mujer de 63 años de edad, con cuadro clínico crónico de un año de evolución caracterizado por diarrea esteatorreica, asociado a episodios de dolor abdominal difuso, tipo cólico, “sensación de bochornos” y enrojecimiento en cara y tronco superior. El abordaje diagnóstico de la diarrea crónica es un reto para los médicos generales y especialistas, más aún, cuando se acompaña de manifestaciones inespecíficas como dolor abdominal y la presencia de “bochornos”. La coexistencia de varios de los anteriores síntomas, obliga a descartar diversas patologías que representan alta morbimortalidad para el paciente. El síndrome de intestino irritable, el feocromocitoma, el hipertiroidismo, el síndrome carcinoide, entre otras, son patologías a excluir en todo caso. El presente artículo pretende brindar el diagnóstico diferencial de las patologías que presentan dichos síntomas, buscando conducir al lector hasta el diagnóstico definitivo de la paciente.
Palabras claves: Tumor carcinoide; síndrome carcinoide maligno; tumor neuroendocrino; cromogranina A; octreótido.
Carcinoid syndrome: case report and literature review
Abstract
A 63-year-old woman reported a chronic clinical evolution of one year characterized by steatorrhea, associated with episodes of diffuse abdominal pain, cramping and “hot flashes” also redness on the face and upper trunk . The diagnostic approach of chronic diarrhea is a challenge for physicians and specialists, especially, when accompanied by nonspecific manifestations such as abdominal pain and the presence of “hot flashes”. The coexistence of several of these symptoms must be ruled various pathologies that represent high morbidity and mortality for the patient. Irritable bowel syndrome, pheochromocytoma, hyperthyroidism, carcinoid syndrome, among others, are conditions to exclude in any case. This article aims to provide the differential diagnosis of the diseases that have these symptoms, seeking to lead the reader to the definitive diagnosis of the patient.
Key words: Carcinoid tumor; malignant carcinoid syndrome; neuroendocrine tumor; chromogranin A; octreotide.
Introducción
La diarrea, el dolor abdominal y el enrojecimiento, rubefacción o flushing, son síntomas frecuentes, aunque inespecíficos. Muchas posibles patologías pueden cursar con estas manifestaciones clínicas, el síndrome de intestino irritable (SII), la enfermedad inflamatoria intestinal (EII), el feocromocitoma, hipertiroidismo y el síndrome carcinoide son algunas de ellas, lo cual destaca la importancia del abordaje correcto del paciente y del conocimiento de algunos puntos claves en el diagnóstico de estas condiciones. La duración de la diarrea es un elemento importante de la anamnesis, pues la diarrea crónica es un reto diagnóstico (1). La coexistencia de rubefacción, diarrea, palpitaciones, cefalea, intolerancia al calor, sibilancias, cambios en la piel o temblor, son factores que pueden sugerir origen endocrino (2). El hipertiroidismo y los tumores carcinoides son causas endocrinas de diarrea crónica (1).
Los tumores carcinoides son raros, representan cerca del 2% de todos los tumores del tracto gastrointestinal (3). La prevalencia global en los Estados Unidos se estima en 1 a 2 casos por cada 100.000 habitantes y la edad de presentación frecuentemente es entre los 50-60 años (4). La localización más frecuente del tumor carcinoide primario es el apéndice, seguido del recto, íleo, pulmones, bronquios y estómago (4). El síndrome carcinoide aparece en menos del 10% de los pacientes con tumor carcinoide y es el único síndrome clínico producido por un tumor endocrino (5); sus manifestaciones clínicas son típicas pero de difícil diagnóstico dada su inespecificidad. La presentación de estos casos en la literatura nacional es escasa, en parte quizá, debido a la dificultad y omisión en el diagnóstico. El presente artículo busca realizar el análisis diferencial del cuadro clínico presentado; pretende llevar al lector a través de un enfoque diagnóstico basado en la semiología y apoyado por resultados de exámenes de laboratorio e imágenes hasta llegar al diagnóstico definitivo, para luego abordar el problema clínico resuelto, ofreciendo una revisión actualizada de la literatura disponible. A través de este, se presenta una patología poco reportada, se describen detalladamente las manifestaciones clínicas, las ayudas paraclínicos, de imágenes y los diagnósticos diferenciales, buscando resaltar la importancia de esta consideración clínica ante síntomas frecuentes pero inespecíficos y procurando brindar herramientas de juicio médico que permitan realizar diagnósticos tempranos, tratamientos específicos y mejora en la calidad de vida del paciente.
Paciente de 63 años de edad, mujer, mestiza, ama de casa. Procede de área urbana. Asiste a consulta médica debido a cuadro clínico de 6 meses de evolución, caracterizado por episodios frecuentes de deposiciones diarreicas, esteatorreicas, abundantes y explosivas, no disentéricas, varios episodios al día, asociado a dolor abdominal tipo cólico, difuso, intermitente, intensidad moderada, no irradiado, acompañado por “sensación de bochornos” y enrojecimiento en cara, parte superior de tórax y brazos. Niega episodios de estreñimiento, tenesmo, hematoquecia y pérdida de peso. Niega identificar factores desencadenantes de las manifestaciones clínicas.
La diarrea es un síntoma frecuente con muchas posibles causas. Puede clasificarse como diarrea aguda, aquella que dura menos de 14 días (6), típicamente autolimitada y de causa infecciosa; y diarrea crónica, más comúnmente de origen no infeccioso. Causas comunes de diarrea crónica incluyen infecciones crónicas en el paciente inmunocomprometido, síndrome de intestino irritable, síndromes de malabsorción, enfermedad inflamatoria intestinal y uso de medicamentos laxantes (7). La duración de la diarrea en esta paciente es clave en el abordaje diagnóstico. El dolor abdominal asociado a diarrea es frecuente en síndrome de intestino irritable y enfermedad inflamatoria intestinal (7) mientras que otros, como los síndromes de malabsorción se caracterizan por diarrea esteatorreica y distensión abdominal, ocurriendo rara vez dolor (8).
La paciente presenta historia personal de hipertensión arterial e hipotiroidismo tratados farmacológicamente (hidroclorotiazida 25 mg/día, enalapril 20 mg/día, levotiroxina 75 mcg/día). Antecedente quirúrgico: histerectomía 3 años atrás debido a prolapso genital. Niega tabaquismo, no consumo de bebidas alcohólicas, niega antecedentes alérgicos e hipersensibilidad conocida a medicamentos. Niega antecedentes familiares de importancia. Revisión por sistemas: niega síntomas. Examen físico: Paciente en aparentes buenas condiciones, en el momento sin enrojecimiento en cara ni tórax, presión arterial 132/84 mmHg, frecuencia cardiaca 78 lpm, frecuencia respiratoria 18 rpm. Cuello sin masas, no ingurgitación yugular, tiroides no dolorosa, sin nódulos. No adenopatías. Sistema cardiorrespiratorio sin alteraciones. Abdomen: blando, depresible, dolor difuso a la palpación profunda, sin masas ni megalias, no signos de irritación peritoneal. Resto de examen físico dentro de parámetros normales. Se solicitan exámenes paraclínicos: Hemograma: hemoglobina 12 g/dl, hematocrito 36%, leucocitos 8200/mm3, diferencial sin alteraciones, plaquetas: 285.000/mm3; creatinina de 1,1 mg/dl y BUN 18 mg/dl; TSH: 2,3 uU/ml.
La paciente es hipotiroidea en tratamiento farmacológico con levotiroxina 75 mcg/día. El hipotiroidismo por sí mismo no se asocia a las manifestaciones gastrointestinales de la paciente. Un exceso en la administración de levotiroxina puede conducir a hipertiroidismo, el cual puede manifestarse como intolerancia al calor y diarrea, ocasionalmente con características esteatorreicas (9); no obstante, el nivel sérico de TSH, revela un adecuado control de la patología, descartando la posibilidad de hipertiroidismo como causa del cuadro clínico.
A la paciente le es solicitado estudios de imágenes, entre estos, ecografía abdominal total la cual es reportada como normal. Adicionalmente se solicita colonoscopia. Dada la historia clínica, los hallazgos al examen físico, los reportes de laboratorio y de ecografía, la paciente recibe un diagnóstico inicial de síndrome de intestino irritable, para lo cual prescriben N-butil bromuro de hioscina y loperamida para manejo ambulatorio.
El síndrome de intestino irritable (SII) es un desorden gastrointestinal funcional, más frecuente en mujeres que en hombres, caracterizado por dolor abdominal recurrente crónico asociado a cambios en el hábito intestinal (10,11). La paciente del caso cumple los criterios diagnósticos para SII (criterios de Roma), sin embargo, la presencia de síntomas de alarma, tales como, inicio de los síntomas después de los 50 años y naturaleza progresiva de los mismos, hace necesario descartar la existencia de neoplasia intraabdominal, particularmente cáncer de colon. Lo anterior requiere la realización de colonoscopia (11). La colonoscopia realizada a la paciente es reportada como negativa para lesiones intraluminales intestinales. Una colonoscopia negativa en pacientes sin historia familiar de cáncer colorectal se asocia a un riesgo extremadamente bajo (1,9%) de desarrollar adenomas avanzados en los próximos 5 años (12). La sensibilidad diagnóstica de la colonoscopia para cáncer y adenomas intestinales es del 89% y 85% respectivamente (13).
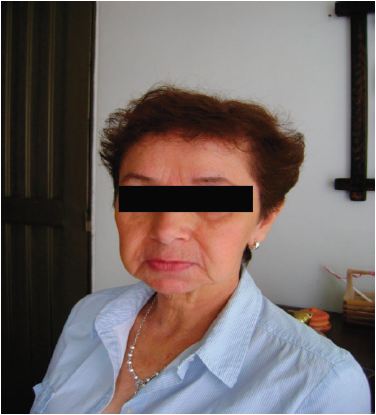
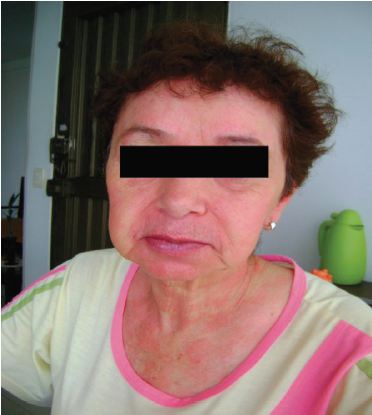
Durante los 6 meses siguientes la paciente recibió tratamiento sintomático, presentando inicialmente leve mejoría. Sin embargo, con el paso de los días, los síntomas gastrointestinales se hicieron cada vez más frecuentes, el flushing aumentó en intensidad y frecuencia (Figura 1) y se asoció en adelante a episodios de sudoración nocturna.
Figura 1. Rubor en paciente con síndrome carcinoide. A la izquierda paciente asintomática, a la derecha rubor en cara y tronco posterior.
La enfermedad inflamatoria intestinal -EII- (Enfermedad de Crohn y Colitis Ulcerativa) tiene un curso intermitente con periodos de exacerbaciones y remisiones, se caracteriza por diarrea crónica, dolor abdominal, fiebre, anorexia y pérdida de peso. La colitis ulcerativa puede además asociarse a episodios de disentería (14). El 15% de diagnósticos de enfermedad inflamatoria intestinal son realizados en personas mayores de 60 años, pero su pico de incidencia se presenta entre los 10 y 40 años de edad, afectando igualmente a hombres y mujeres (14). No existen descripciones en la literatura que asocien la EII con las manifestaciones dermatológicas descritas en la paciente.
La enfermedad celiaca, es una enfermedad autoinmune que se da en personas genéticamente predispuestas; entre la población adulta, es 2 a 3 veces más frecuente en mujeres, se caracteriza por diarrea acompañada en algunas ocasiones de dolor o malestar abdominal y en raras oportunidades de estreñimiento, pérdida de peso, anemia ferropénica u osteoporosis (15). La enfermedad celiaca típicamente es desencadenada por el consumo de gluten (15); característica no descrita como desencadenante por la paciente
La coexistencia de rubefacción, diarrea, palpitaciones, cefalea, intolerancia al calor, sibilancias, cambios en la piel o temblor, son factores que pueden sugerir una causa endocrina (2). El carcinoma medular de tiroides representa el 5 al 8% de los tumores tiroideos, usualmente se manifiesta como nódulos tiroideos palpables y la presencia de flushing y diarrea hacen sospechar esta entidad. La presencia de una tiroides normal durante la palpación del cuello, hacen que esta posibilidad diagnóstica sea poco probable. (16).
La sudoración y el enrojecimiento son síntomas típicos en el feocromocitoma, el hipertiroidismo y el síndrome carcinoide (SC) (2). El feocromocitoma, un tumor de glándulas suprarrenales, representa el 0,3 a 1,9% de las causas secundarias de HTA; su distribución en frecuencia es igual en hombres y mujeres (17). La edad media de diagnóstico es a los 40 años; su curso clínico característicamente se manifiesta con la triada de episodios paroxísticos de cefalea (80%), diaforesis (57%) y palpitaciones (64%) en un paciente con hipertensión sostenida o intermitente. La cefalea, la hipertensión y las palpitaciones son más frecuentes (87%) que los episodios de sudoración (37%) (17-19). La triada está presente hasta en el 40% de los pacientes y tiene una sensibilidad del 90,9% y especificidad del 93,8% para el diagnóstico en pacientes hipertensos. La diarrea no es manifestación de esta entidad (17-19).
En el síndrome carcinoide la rubefacción cutánea es el síntoma típico y aparece en el 84% de los pacientes. Otros síntomas comunes son la diarrea acuosa y las sibilancias. La rubefacción, raramente aparece aislada y por lo tanto la hipótesis diagnóstica de un síndrome carcinoide puede diferirse si la rubefacción se presenta como única manifestación (2). Con excepción del carcinoide, el flushing debido a tumores es raro y tiende a ocurrir en etapas avanzadas (20).
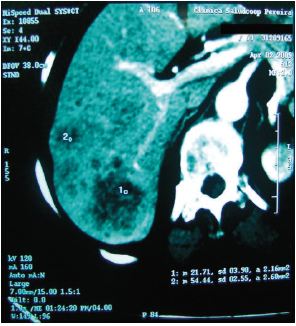
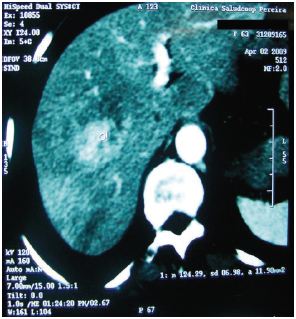
En los meses siguientes la sintomatología persistió, fue progresiva y la paciente presentó pérdida de peso de aproximadamente 7 kg, además, afección de su calidad de vida. Las anteriores razones, llevaron a que se decidiera realizar una tomografía axial computarizada (TAC) de abdomen con doble contraste. Su reporte reveló la presencia de lesiones hepáticas nodulares metastásicas de origen no determinado, sin hallazgos adicionales en otros órganos (Figura 2).
Figura 2. TAC contrastada. Presencia de lesiones hipervasculares de predominio en lóbulo hepático derecho.
Los hallazgos en la TAC contrastada de abdomen, hacen poco probable el diagnóstico de feocromocitoma, dada la ausencia de lesiones tumorales a nivel de las glándulas suprarrenales. El 85% de los feocromocitomas se ubican en las glándulas suprarrenales y más del 98% se encontrarán en el abdomen. La TAC tiene una sensibilidad cercana al 90% para feocromocitoma adrenal y aumenta a 95% para tumores mayores de 0,5 cm de diámetro, sin embargo son menos sensibles (<91%) para detectar metástasis, recurrencias y tumores extra adrenales (21).
La presencia de lesiones hepáticas metastásicas, requiere estudios complementarios para identificar el sitio de la lesión primaria, pues casi todos los tumores sólidos pueden hacer metástasis hepáticas, entre estos, los cánceres primarios del aparato gastrointestinal (colon, páncreas, estómago, tumores neuroendocrinos), seno, pulmón, riñón, glándula adrenal, ovario, útero, melanoma y sarcomas (22).
Después de 12 meses de evolución la paciente es derivada a consulta de cirugía general, al momento de la evaluación manifiesta progresión y empeoramiento de los síntomas, pérdida de aproximadamente 10 kg de peso y gran deterioro en su calidad de vida. Refiere múltiples episodios de diarrea al día y flushing. Debido al cuadro clínico, los hallazgos al examen físico, de laboratorio e imágenes, se solicitan estudios de extensión: Radiografía de tórax, antígeno carcinoembrionario (CEA), alfa feto proteína (AFP), Ca 19.9 y mamografía bilateral. Todos los anteriores estudios fueron reportados sin alteraciones.
El síndrome carcinoide es una de las entidades más importantes en el diagnostico diferencial de flushing, debido a la naturaleza maligna del tumor carcinoide y la relativa alta mortalidad. Por tanto, el SC debe ser sospechado y descartado en pacientes quienes presenten flushing. Clásicamente el SC presenta la triada de flushing, hipermotilidad intestinal (cólico abdominal y diarrea) y falla cardiaca del lado derecho debido a enfermedad valvular (20). El 95% de pacientes con SC tienen flushing en algún momento de la enfermedad, haciendo de este el signo clínico más frecuente (20). El SC ocurre en aproximadamente el 10% de pacientes con tumor carcinoide (20).
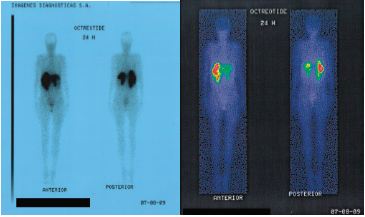
Ante la sospecha de síndrome carcinoide se procede a solicitar Cromogranina A en sangre, reportando 655.60 ng/ml (valor referencia menor 36.5 ng/mL), Ácido 5-hidroxi-indolacético urinario: 164.00 mg/24h (valor referencia 2 - 10 mg/24h), gammagrafía marcada con octreótido (Octreoscan) la cual reporta múltiples lesiones metastásicas en ambos lóbulos hepáticos y dos metástasis ganglionares de probable localización retroperitoneal derecha (Figura 3).
Figura 3. Gammagrafía con octreotide. Múltiples lesiones hepáticas.
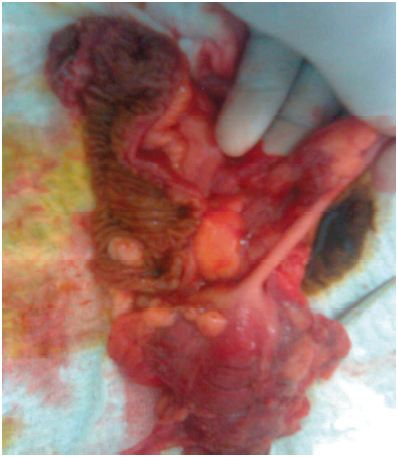
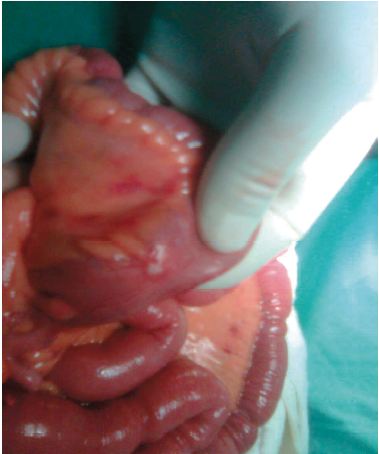
El estudio histopatológico de las lesiones hepáticas revela metástasis de tumor neuroendocrino. Posteriormente se realiza exploración quirúrgica mediante laparotomía, logrando identificar el sitio del tumor primario en intestino delgado (Figura 4).
Figura 4. Identificación del tumor primario mediante exploración quirúrgica. Asas de íleon con engrosamiento difuso de la pared, color ámbar y tumoraciones pequeñas a nivel de la capa serosa (iziquierda). Asas con importante hiperemia y tumoración con compromiso de la capa serosa (derecha).
Los tumores neuroendocrinos y el síndrome carcinoide son patologías raras que exigen un alto grado de sospecha clínica, especialmente en pacientes con síntomas abdominales recurrentes o tras el descubrimiento de hepatomegalia o metástasis hepáticas (23).
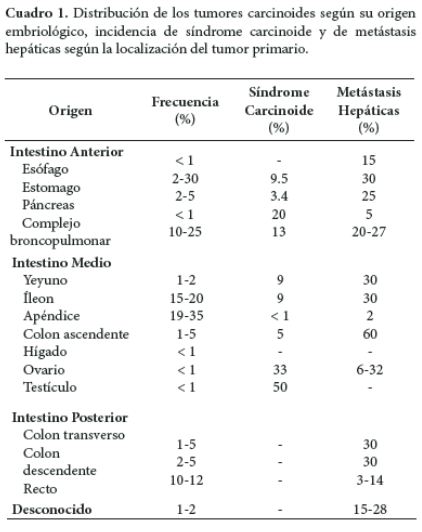
Los tumores carcinoides se derivan de la proliferación neoplásica de las células enterocromafines. Ésta denominación, si bien, es muy utilizada, ha sido reemplazada en los últimos años por tumor neuroendocrino del tracto gastroenteropancreático (24). El síndrome carcinoide aparece en menos del 10% de los pacientes con tumor carcinoide y es el resultado de la metástasis de un tumor neuroendocrino derivado de intestino medio (24). Los tumores carcinoides se clasifican según su derivación a partir de las divisiones embrionarias del intestino primitivo (4, 25-27); en caso de metástasis, describe la ubicación del tumor primario (26). El Cuadro 1, relaciona la distribución de los tumores carcinoides según su origen embriológico, incidencia de síndrome carcinoide y de metástasis hepáticas según la localización del tumor primario.
Los carcinoides usualmente son asintomáticos y se hallan incidentalmente durante una cirugía abdominal (4). Si hay presencia de síntomas, estos son vagos e inespecíficos. El tiempo medio desde la aparición de los síntomas hasta el diagnóstico es mayor a nueve años. (3, 4, 28). Las manifestaciones clínicas dependen de la localización anatómica, tamaño del tumor y la presencia de metástasis (4, 29). Pueden presentarse como un cuadro de obstrucción intestinal, dolor abdominal, náuseas, vómito, colestasis, manifestarse por las metástasis hepáticas, o como un síndrome típico por hipersecreción hormonal (3, 24, 28).
El síndrome carcinoide resulta de un tumor neuroendocrino metastásico derivado de intestino medio, y es el único síndrome clínico producido por un tumor endocrino. Los carcinoides del intestino anterior pueden producir un síndrome clínico característico conocido como carcinoide atípico, sus manifestaciones dependerán del perfil exacto y el metabolismo de las hormonas secretadas; los del intestino posterior generalmente son no funcionantes (24, 25).
Los tumores carcinoides pueden liberar serotonina, histamina, prostaglandinas, kalicreina, bradicininas, sustancia P, gastrina, corticotropina, taquicininas y enolasa neuronal específica (4, 24, 30). La presentación clínica depende de la combinación de sustancias secretadas. El ácido 5-hidroxiindolacetico (5-HIAA) es un metabolito de la serotonina que se excreta en orina (4, 24). Entre las hormonas secretadas por las células tumorales se encuentra la cromogranina A y otros péptidos relacionados, que podrían servir como marcadores tumorales en pacientes con enfermedad metastásica (25).
El síndrome carcinoide clásico o típico ocurre en menos del 10% de los pacientes, es causado por la liberación de serotonina y otros compuestos vasoactivos que alcanzan la circulación sistémica cuando se han desarrollado metástasis hepáticas o cuando el tumor primario está en una localización atípica como bronquios u ovarios (24, 27). Sus manifestaciones incluyen rubor (más frecuente en cara, cuello y parte superior del tórax), diarrea, dolor y cólicos abdominales (3, 4, 24, 28, 30); los cuales pueden variar en intensidad y cronicidad, así como ser de presentación paroxística y responder a factores desencadenantes como ejercicio, estrés, alcohol y alimentos ricos en serotonina (queso, café, chocolate) (24).
Manifestaciones menos frecuentes incluyen taquicardia, hipotensión, broncoespasmo, epífora, diaforesis profusa, telangiectasias, y signos de pelagra (demencia, dermatitis y diarrea) (3, 4, 24). Otras posibles manifestaciones son hepatomegalia, asma, enfermedad valvular cardiaca derecha (las válvulas izquierdas generalmente se ven menos afectadas por el metabolismo de la serotonina en los pulmones) (3,4, 24).
No se tiene certeza de la sustancia responsable del rubor o flushing; la diarrea parece ser causada por el exceso de serotonina en la circulación, y se caracteriza por ser episódica, acuosa y explosiva (4, 27). El broncoespasmo podría estar mediado por la serotonina y la bradicinina. La enfermedad valvular cardiaca del lado derecho se presume es causada por niveles séricos elevados y prolongados de serotonina y es por tanto una complicación tardía (27). El Cuadro 2, relaciona las manifestaciones clínicas, características y mediadores involucrados en el SC.
En caso de sospechar síndrome carcinoide, el diagnóstico debe confirmarse con exámenes paraclínicos, que además buscan determinar la presencia de metástasis y la ubicación del tumor primario; hacen parte de estos estudios análisis de orina y suero, estudios de imágenes y medicina nuclear (31).
El nivel de 5-HIAA en orina de 24 horas, es la prueba más usada en el estudio de los carcinoides, tiene una sensibilidad del 73% y especificidad de 100% para el diagnóstico de tumor carcinoide (32). Este metabolito no se eleva en carcinoide atípico, no obstante, puede elevarse en otras condiciones como esprue tropical, enfermedad de Whipple, consumo de alimentos ricos en serotonina (plátanos, piña, nueces y aguacates) y medicamentos como fluoxetina, acetaminofén, o salicilatos (33).
La cromogranina A es una glicoproteína secretada por estas neoplasias, algunos estudios le avalan sensibilidad del 80% y especificidad del 95% en el diagnóstico de esta patología (34). Es un marcador pronóstico y de remisión tumoral; altos niveles plasmáticos se asocian a pobre pronóstico (35).
Los estudios imagenológicos en tumor carcinoide son relativos al origen y al grado de diferenciación neoplásica al tiempo del diagnóstico (36). La tomografía axial es útil para valorar extensión mesentérica y la presencia de metástasis hepáticas, pero falla en el hallazgo del tumor primario en un 40-60%, al igual que la ultrasonografía, pasando por alto lesiones menores a 2 cm de diámetro (37).
La gammagrafía con octreótido (análogo de somatostatina) marcado con indio-111, también llamado octreoscan, es la prueba de elección para enfermedad metastásica, su sensibilidad es del 61-96% (3841). En casos donde no se encuentra el tumor primario por estos métodos, está indicada la exploración quirúrgica para identificarlo. Aproximadamente el 11%-14% de los tumores neuroendocrinos, tienen metastasis con un tumor primario desconocido, para lo cual se hace necesario la exploracion quirurgica por medio de laparoscopia o laparotomia, buscando identificar y resecar el tumor, con el fin de tratar y prevenir complicaciones locales como dolor, obstruccion, isquemia o perforacion. Los tumores primarios son frecuentemente encontrados en intestino delgado y usualmente son pequeños y multifocales, (42).

Las decisiones de tratamiento en el síndrome carcinoide son situacionales e incluyen: cambios en el estilo de vida, evitando condiciones que puedan desencadenar flushing; uso de complementos alimenticios con nicotinamida, tratamiento de la insuficiencia cardiaca con diuréticos, de las sibilancias con bronco dilatadores y control de la diarrea con agentes como la loperamida o el difenoxilato. Si persiste la sintomatología, los antagonistas del receptor de serotonina (ondansetrón) o los análogos de la somatostatina son los medicamentos indicados (43).
En adición a la terapia sistémica, la terapia de ablación por radiofrecuencia es usada para disminuir la secreción peptídica del tumor y la carga tumoral (44). La quimioterapia debe emplearse sólo en forma paliativa en pacientes sintomáticos (45).
La resección de las lesiones primarias es el tratamiento de elección para los tumores neuroendocrinos; sin embargo, debido a su curso asintomático, la mayor parte de los pacientes (75%) presenta lesiones metastásicas al momento de consulta (46). En pacientes con metástasis hepáticas, se ha encontrado que su supervivencia a 5 años se reduce de 90 a 40% y en caso de que el tumor desarrollado sea funcional, la totalidad de pacientes terminará presentando el síndrome (47).
La reducción completa del cáncer debe ser considerada en todos los casos, la hepatectomía prolonga la supervivencia (de 41% a un 86%), con bajos porcentajes de mortalidad derivada del procedimiento (0-6,7%) (47). En pacientes que desarrollan el síndrome carcinoide resistente a la terapia médica, se puede acudir la resección incompleta de las metástasis hepáticas, lo cual reduce la sintomatología y detiene el progreso hacia enfermedad cardiaca carcinoide. Ya en pacientes con tumores irresecables, el trasplante hepático ha mostrado aumentar la supervivencia a 5 años hasta en el 77% de pacientes (47). La cirugía estará indicada en pacientes en buen estado general y con enfermedad limitada (v.g. primario con o sin metástasis linfáticas regionales), además de pacientes con metástasis hepáticas y enfermedad potencialmente resecable (48).
Es importante destacar que si bien el síndrome carcinoide no es una entidad frecuente, es fundamental el conocimiento de sus manifestaciones clínicas, debido a su naturaleza maligna y relativa alta mortalidad. La paciente del caso aquí presentado, muestra las manifestaciones típicas de un síndrome carcinoide, el flushing o rubor, la diarrea y el dolor abdominal tipo cólico son algunas de estas. El síndrome carcinoide es una de las entidades más importantes en el diagnóstico diferencial del flushing. El diagnóstico en todo caso debe partir desde la sospecha clínica, no obstante, las ayudas diagnósticas a utilizar siempre ofrecen información de trascendental importancia, algunas de ellas incluyen: análisis de orina (ácido 5-hidroxi-indolacético), suero (cromogranina A), estudios de imágenes (ecografía y/o TAC) y medicina nuclear (gammagrafia marcada con octreótido), e incluso la exploración quirúrgica para aquellos casos en donde no se identifica el tumor primario. En pacientes quienes presentan metástasis hepáticas la mortalidad es bastante elevada, y en caso de que el tumor sea funcional, la totalidad de pacientes desarrollará el síndrome.
Finalmente, debido a que el síndrome carcinoide suelen presentarse con manifestaciones clínicas inespecíficas y numerosos procedimientos de investigación se realizan antes de establecer el diagnóstico correcto, es fundamental conocer la presentación y cuadro clínico de ésta entidad, pues el adecuado estudio y enfoque del paciente permitirá realizar la aproximación diagnóstica y uso de los métodos y técnicas diagnósticas apropiadas de acuerdo a la presentación clínica particular, así como de las opciones terapéuticas disponibles en la actualidad.
Los autores declaran no tener conflictos de interés.
1. Juckett G, Trivedi R. Evaluation of Chronic Diarrhea. Am Fam Physician 2011;84(10):1119 -1126. [ Links ]
2. Buitrago F, Alejandre C, Marroyo J. Abordaje de la sudoración nocturna. Med Contin Aten Prim 2010;17(9):570-577. [ Links ]
3. Massironi S, Sciola V, Peracchi M, Ciafardini C, Spampatti M, Conte D. Neuroendocrine tumors of the gastro-entero-pancreatic System. J Gastroenterol 2008;14(35):5377-5384. [ Links ]
4. Robertson R, Geiger W, Davis N. Carcinoid Tumors. Am Fam Physician 2006;74(3):429-434. [ Links ]
5. Arnold R. Introduction: Definition, historical aspects, classification, staging, prognosis and therapeutic options Clinical Gastroenterology and Hepatology 2005;19:491-505. [ Links ]
6. Thielman N, Guerrant R. Acute Infectious Diarrhea. N Engl J Med 2004;350:38-47. [ Links ]
7. Kapoor R, Moseley R, Kapoor J, Crapo L, Saint S. Needle in a Haystack. N Engl J Med 2009;360(6):616-621. [ Links ]
8. Hodgson H, Epstein O. Malabsorption. Medicine 2007;35:220-225. [ Links ]
9. Cooper D. Hyperthyroidism. Lancet 2003;362(9382):459-468. [ Links ]
10. Wilkins T, Pepitonec C, Alex B, Schade RR. Diagnosis and Management oflBS in Adults. Am Fam Physician 2012;86(5):419-426. [ Links ]
11. Mayer E. Irritable Bowel Syndrome. N Engl J Med 2008;358(16):1692-1699. [ Links ]
12. Imperiale T, Glowinski E, Lin-Cooper C, Larkin G, Rogge J, Ransohoff D. Five-year risk of colorectal neoplasia after negative screening colonoscopy. N Engl J Med 2008;359(12):1218-1224. [ Links ]
13. Hassan C, Di Giulio E, Marmo E, Zullo A, Annibale B. Appropriateness of the indication for colonoscopy: systematic review and meta-analysis. J Gastrointestin Liver Dis 2011;20(3):279-286. [ Links ]
14. Bianchi M. Inflammatory bowel diseases, celiac disease, and bone. Arch Biochem Biophys 2010;503(1):54-65. [ Links ]
15. Green P, Cellier C. Celiac Disease. N Engl J Med 2007;357(17):1731-1743. [ Links ]
16. Pacini F, Castagna M, Cipri C, Schlumberger M. Medullary Thyroid Carcinoma. Clin Oncol 2010;22(6):475-485. [ Links ]
17. Ripodas B, Arillo A, Murie M, Garcia D. Pheochromocytoma A case report. An Sist Sanit Navar 2012;35(1):121-125. [ Links ]
18. Subramaniam R. Pheochromocytoma curret concepts in diagnosis and management. Trends in anaesthesia and critical care 2011;1(2):104-110. [ Links ]
19. Zuber S, Kantorovich V, Pacak K. Hypertension in pheochromocytoma: Characteristics and treatment. Endocrinol Metab Clin North Am 2011;40(2): 295-311. [ Links ]
20. Izikson L, English J, Zirwas M. The flushing patient: differential diagnosis, workup, and treatment. J Am Acad Dermatol 2006;55(2):193-208. [ Links ]
21. Fitzgeral PA. Endocrine disorders In: McPhee FJ, Papadakis MA. Current Medical Diagnosis & Treatment. 15 ed. USA: Mc Graw Hill; 2011. [ Links ]
22. Jarnagin WR. Liver & portal venous system In: Doherty GM. Current Diagnosis and Treatment: Surgery. 13 ed. USA: Mc Graw Hill; 2010. [ Links ]
23. Arnold R, Rinke A, Schmidt Ch, Hofbauer L. Endocrine tumors of the gastrointestinal tract: Chemotherapy. Best Pract Res Clin Gastroenterol 2005;4:549-556. [ Links ]
24. Van der Lely A. de Herder W Carcinoid syndrome: diagnosis and medical management. Arq Bras Endocrinol Metab 2005;49(5):850-860. [ Links ]
25. Arnold R. Introduction: Definition, historical aspects, classification, staging, prognosis and therapeutic options. Best Pract Res Clin Gastroenterol 2005;19(4):491-505. [ Links ]
26. Kulke M, Mayer R. Carcinoid tumors. N Engl J Med 1999;340:858-868. [ Links ]
27. Woodside K, Townsend C, Mark Evers B. Current Management of Gastrointestinal Carcinoid Tumors. J Gastrointest Surg 2004;8(6):742-756. [ Links ]
28. Gaitán M., Cadena M., Vergara A. Tumor neuroendocrino del intestino delgado. Reporte de un caso singular. Rev Colomb Cir 2005;20(4):222-230. [ Links ]
29. Townsend A, Price T, Yeend S, Pittman K, Patterson K, Luke C. Metastatic Carcinoid Tumor Changing Patterns of Care Over Two Decades. J Clin Gastroenterol 2010;44(3):195-199. [ Links ]
30. Lazarte R, Poniachik J, Smok G, Contreras J, Gutiérrez L, Csendes A. Tumores neuroendocrinos gástricos: presentación clínica, endoscópica y alternativas de tratamiento. Rev Méd Chile 2002;130(9):985-992. [ Links ]
31. Pinchot S, Holen K, Sippel R, Chen H. Carcinoid Tumors. Oncologist 2008;13(12):1255-1269. [ Links ]
32. Modlin I, Kidd M, Latich I, Zikusoka M, Shapiro M. Current status of gastrointestinal carcinoids. Gastroenterology 2005;128:1717-1751. [ Links ]
33. Roberts L, Anthony L, Oates J. Disorders of vasodilator hormones; carcinoid syndrome and mastocytosis. In: Williams RH, Wilson JD, eds. Williams Textbook of Endocrinology. 9th ed. Philadelphia, Pa.: Saunders, 1998:1711-1720. [ Links ]
34. Nobels F, Kwekkeboom D, Coopmans W, Schoenmakers C, Lindemans J, De Herder W, et al. Chromogranin A as serum marker for neuroendocrine neoplasia: comparison with neuron-specific enolase and the alpha-subunit of glycoprotein hormones. J Clin Endocrinol Metab 1997;82:2622-2630. [ Links ]
35. Seregni E, Ferrari L, Bajetta E, Martinetti A, Bombardieri E. Clinical significance of blood chromogranin A measurement in neuroendocrine tumours. Ann Oncol 2001;12:69-72. [ Links ]
36. Pasieka J, McKinnon J, Kinnear S, Yelle C, Numerow L, Paterson A, et al. Carcinoid syndrome symposium on treatment modalities for gastrointestinal carcinoid tumours: symposium summary. Can J Surg 2001;44:25-32. [ Links ]
37. Galiber A, Reading C, Charboneau J, Sheedy P, James E, Gorman B, et al. Localization of pancreatic insulinoma: comparison of pre-and intraoperative US with CT and angiography. Radiology 1988;166:405-408. [ Links ]
38. Ramage J, Davies A, Ardill J, Bax N, Caplin M, Grossman A, et al. Guidelines for the management of gastroenteropancreatic neuroendocrine (including carcinoid) tumours. Gut 2005;54(4):1-16. [ Links ]
39. Jensen R, Gibril F, Termanini B. Definition of the role of somatostatin receptor scintigraphy in gastrointestinal neuroendocrine tumor localization. Yale J Biol Med 1997;70:481-500. [ Links ]
40. Chiti A, Briganti V, Fanti S, Monetti N, Masi R, Bombardieri E. Results and potential of somatostatin receptor imaging in gastroenteropancreatic tract tumours. Q J Nucl Med 2000;44:42-49. [ Links ]
41. Miederer M, Weber M, Fottner C. Molecular Imaging of Gastroenteropancreatic Neuroendocrine Tumors. Gastroenterol Clin N Am 2010;39:923-935. [ Links ]
42. Wang S, Parekh J, Zuraek M, Venook A, Bergsland E, Warren R, et al. Identification of unknown primary tumors in patients with neuroendocrine liver metastases. Arch Surg 2010;145:276-280. [ Links ]
43. Reubi J. Peptide receptors as molecular targets for cancer diagnosis and therapy. Endocrinol Rev 2003;24:389-427. [ Links ]
44. Lips C, Lentjes E, Hoppener J. The spectrum of carcinoid tumours and carcinoid syndromes. Acta Bioquím. Clín. Latinoam 2004;38(1):47-59. [ Links ]
45. Landry C, Scoggins C, McMasters K, et al. Management of hepatic metastases of gastrointestinal carcinoid tumors. J Surg Oncol 2008;97:253-258. [ Links ]
46. Boleslawski E, Dharancy S, Truant S, Pruvot FR. Surgical management of liver metastases from gastrointestinal endocrine tumors. Gastroenterol Clin Biol 2010;34(4-5):274-282. [ Links ]
47. Que F, Sarmiento J, Nagorney D, et al. Hepatic surgery for metastatic gastrointestinal carcinoid tumors. Cancer Control 2002;9(1):67-79. [ Links ]
48. Ramage J, Davies A, et al. Guidelines for the management of gastroenteropancreatic neuroendocrine (including carcinoid) tumours. Gut 2005;54(4):1-16. [ Links ]
Rev. Méd. Risaralda 2014; 20 (1): 60-67
