










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Médica de Risaralda
Print version ISSN 0122-0667
Revista médica Risaralda vol.20 no.2 Pereira July/Dec. 2014
Reporte de Caso
Trastornos de la migración neuronal: un caso de lisencefalia
Julio Curiel,1,2 Martin Alonso Olaya Ordoñez,2* Wilman Orlando Ortiz Fonseca,2 Manuel Leonardo Lozano Diaz.2
1 Servicio de Neuropediatría, Hospital Universitario Fernando Troconis, Santa Marta, Magdalena, Colombia.
2 Programa de Medicina, Universidad del Magdalena, Santa Marta, Magdalena, Colombia.
* Correo electrónico: martinol_01@hotmail.com
Fecha de Recepción: 25-01-2014.
Fecha de Solicitud de Correcciones: 03-08-2014.
Fecha de Aceptación: 18-09-2014.
Fecha de Publicación Online: 18-09-2014.
Resumen
Las malformaciones del desarrollo cortical originan un grupo de patologías, que cursan con diversas manifestaciones las cuales marcan un grado de minusvalía significativo en quienes las padecen. La Lisencefalia es una de las alteraciones de la migración neuronal caracterizada por una corteza cerebral de superficie lisa o con pocas circunvoluciones. Este trastorno está frecuentemente asociado a epilepsias de difícil manejo. Se presenta el caso de un paciente masculino de 18 meses de edad con diagnóstico de Lisencefalia quien cursa con convulsiones tónicas generalizadas desde los 8 meses de edad. Se le realizó TAC de cráneo simple y resonancia magnética de cerebro para confirmar el diagnóstico y se dio manejo farmacológico a las convulsiones.
Palabras clave: lisencefalia; malformaciones del desarrollo cortical; paquigiria; epilepsia.
Neuronal migration disorders: a lissencephaly case Abstract
Malformations of cortical development originate a group of pathologies that involve diverse manifestations that mark a significant degree of disability in those who suffer from. The Lissencephaly is one of alterations in neuronal migration characterized by a smooth surface or with few convolutions in the cerebral cortex. This disorder is often associated with difficult management Epilepsies. We report the case of a male patient from 18 months of age with a diagnosis of Lissencephaly, who presents generalized tonic seizures from 8 months of age. Was performed a simple skull CT and brain magnetic resonance to confirm the diagnosis and the pharmacological management was given to seizures.
Key Words: lissencephaly; malformations of cortical development; pachygyria; epilepsy.
Introducción
Las alteraciones del desarrollo del sistema nervioso central son una causa de minusvalía muy importante, el 3% de los neonatos presentan alguna malformación del sistema nervioso; de este grupo, el 70% no son compatibles con la vida (1). De los trastornos de migración neuronal, la Lisencefalia con Síndrome de Miller-Dieker, tiene una incidencia de 11,7 casos por millón de recién nacidos (1/85.470) pero la prevalencia de formas más leves es desconocida (2).
Las causas son heterogéneas, aproximadamente el 80% de los casos se deben a errores genéticos, alteraciones de los cromosomas 17, cromosoma X y en el gen de la reelina, ubicado en el cromosoma 7. También cabe mencionar que existen factores ambientales asociados como la exposición materna al etanol, ácido retinoico, metilmercurio y radiación, insecticidas o infecciones prenatales virales como citomegalovirus (2-3).
Una detección temprana de cualquier alteración en el desarrollo fetal o el conocimiento de los factores de riesgo que predisponen a dichas alteraciones podría significar la diferencia entre un niño sano y otro con secuelas asociadas a retraso mental profundo y epilepsias de difícil control (2). Cuando hablamos de epilepsias de difícil control nos referimos a aquellas que están relacionadas con factores clínicos tales como el inicio temprano de las crisis convulsivas (<2,9 años), resonancia magnética cerebral con hallazgos anormales y origen sintomático o secundario (4).
Estas anomalías son muy limitantes afectando no solo a quien la padece sino también a quienes los rodean; especialmente a las madres, y que son causa frecuente de rupturas matrimoniales, es importante dar a conocer un poco más sobre el tema, esperando que sea de gran utilidad para el clínico quien deberá estar entrenado para diagnosticar y tratar el curso sintomático de estos trastornos.
Reporte de Caso
Paciente masculino de 18 meses de edad, producto de la primera de gestación de madre menor de 16 años de edad, embarazo no controlado, sin pruebas serológicas ni administración de micronutrientes durante el embarazo. Refiere la madre que solo hasta cuatro meses después de haber quedado en estado de embarazo, se enteró de su condición al realizarse una ecografía. Como dato relevante está la consanguinidad de los padres (primos) y la exposición a insecticidas en el primer trimestre de embarazo por parte de la madre. El paciente fue obtenido por cesárea a las 36 semanas de gestación con sufrimiento fetal agudo.
Al momento del nacimiento presento Apgar de 4 y de 10 al minuto, no requirió maniobras de reanimación; su peso fue de 2200 gr, talla de 48 cms y perímetro cefálico de 30 cms; fue internado en la Unidad de Cuidados Intensivos Neonatal durante 7 días, desde donde egresa con diagnóstico de microcefalia y pie equino varo bilateral.
Cursó con retraso en el desarrollo psicomotor desde el nacimiento, el paciente no desarrollo sonrisa social no es capaz de seguir objetos con la vista, no es capaz de sentarse, a esto se asocian convulsiones tónicas generalizadas de aproximadamente 1 minuto de duración en número de más de 10 al día desde los ocho meses de edad.
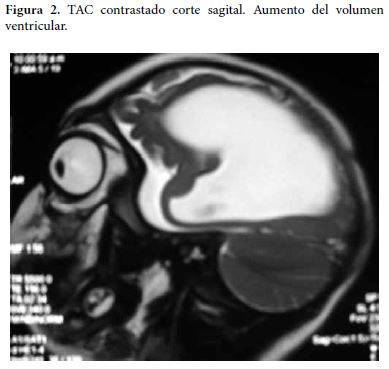
Al examen físico actual se encuentra paciente con peso de 10 kilos, fontanelas cerradas, perímetro cefálico de 41 cms, hiperextensión de cuello y tronco, artrogriposis de las articulaciones de rodillas y tobillos y deformidad en varo de los pies. Se le realiza TAC de cráneo simple y contrastado que muestra dilatación del sistema ventricular con atrofia marcada del parénquima cerebral (Figura 1 y 2).

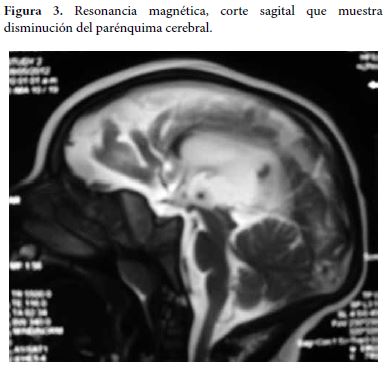
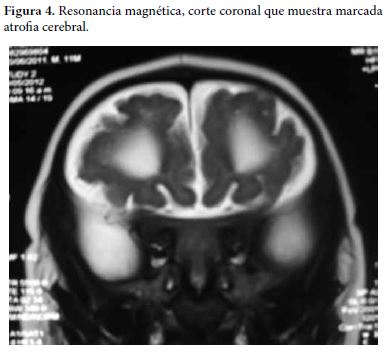
Se realiza una resonancia magnética de cerebro en donde se evidencia dilatación del sistema ventricular con circunvoluciones cerebrales escazas, además de agenesia del cuerpo calloso, considerando trastorno de migración neuronal (Figura 3 y 4).
Inicialmente se manejaron las crisis convulsivas con Fenobarbital vía oral a razón de 6 mg/Kg/día, pero al no obtener el control de las mismas se utilizó Carbamazepina vía oral a dosis de 15 mg/Kg/día terapia que hasta el momento ha resultado eficaz.
Discusión
Luego de completarse el cierre del tubo neural al final de la tercera semana del desarrollo embrionario y después que se han formado las vesículas telencefálicas (5), se inicia el proceso de migración celular, ya que la mayoría de las neuronas que conforman la corteza nacen en lugares distintos al lugar donde finalmente se establecen (5-6).
Se sabe que al igual que otras células, las neuronas migran, siguiendo tres pasos esquemáticos:
1) En primer lugar, la célula extiende sus bordes anteriores precedida por proyecciones citoplasmáticas (filopodios y lamelopodios) que exploran el microentorno (5). Este proceso de extensión del borde celular se lleva a cabo después de la integración de las señales atractivas y repulsivas generadas en la membrana plasmática.
2) En segundo lugar, la migración celular requiere que el núcleo se mueva en dirección de la extensión del borde, una etapa conocida como nucleocinesis (5).
3) El tercer evento en la migración neuronal corresponde a la retracción de los bordes o procesos posteriores (retracción de los procesos de rastreo) (6). De esta manera, las células se ubican en las diferentes capas de la corteza y cualquier alteración (genética, cromosómica o ambiental) durante este proceso podría ocasionar importantes malformaciones cerebrales (7).
En este caso particular, La Lisencefalia es una de las alteraciones de la migración neuronal, que implica patológicamente agiria (pérdida total de circunvoluciones) y paquigiria (disminución de circunvoluciones). Radiológicamente se clasifica en seis grados, dependiendo de las cantidades relativas de agiria y paquigiria, y la presencia o ausencia de heterotopía (8).
Sin embargo, la clasificación más utilizada y revisada de Lisencefalia incluye la llamada Lisencefalia tipo I en la que el cortex está constituido por cuatro capas en vez de seis como debería ser en condiciones normales y Lisencefalia tipo II en la que anátomo-patológicamente se detecta una mezcla de agiria, paquigiria y micropoligiria.
Se acepta también una subclasificación de Lisencefalia tipo I, la Lisencefalia aislada (ISL) y el síndrome de Miller-Dieker (SMD) (910). En ambos subtipos clínicamente aparece una epilepsia de difícil manejo con morfología de espasmos infantiles con o sin registro EEG hipsarrítmico y tienen un origen genético que implica una mutación en el cromosoma 17p13.3 en el gen LIS1 que codifica una subunidad del factor acetil hidrolaxa que se encarga de activar el desarrollo del cortex y de activar la molécula neurorreguladora PAF (Platelet-Activating Factor).
El SMD es un síndrome que afecta genes contiguos a diferencia de ISL que solo afecta un gen (9-11), lo cual genera las características faciales propias de los niños que padecen esta enfermedad (12-14).
La Lisencefalia también puede estar asociada a otras malformaciones corporales que hacen difícil la supervivencia del individuo. Se ha descrito también casos de enanismo con microcefalia osteodisplásica primordial asociada a Lisencefalia cuya expectativa de vida no supera los 30 años de edad (15).
Al ser un grupo heterogéneo de factores que contribuyen a que se presenten los trastornos de la migración neuronal y al ser este desorden tan incapacitante, tomar medidas para detectar estas anomalías debería ser una intervención de tamizaje en el curso de un embarazo.
En la etapa neonatal se hace también necesaria la identificación de los niños que posean esa condición, porque con el aumento de la natalidad también aumenta su incidencia; además del evidente retraso neurológico y la posible aparición de convulsiones de difícil manejo radiológicamente se observa una superficie cerebral lisa, un manto cortical engrosado e indicios microscópicos de migración neuronal incompleta, (15-19).
Los cambios en la corteza cerebral ocurren durante la primera mitad del embarazo, donde ante la presencia de eventos nocivos para el feto, este es vulnerable y puede padecer algún defecto en el desarrollo del SNC. Dichas anomalías están ampliamente documentadas en varios estudios epidemiológicos, los cuales demuestran una relación directa entre malformaciones congénitas de todo tipo y el contacto con pesticidas (20).
Teniendo en cuenta esto podemos concluir que durante el desarrollo embriológico de nuestro paciente existió una noxa en el inicio de la gestación que intervino en el proceso de migración neuronal como fue la exposición de la madre a insecticidas durante el primer trimestre de embarazo y la no asistencia a controles prenatales que hubiesen hecho posible detectar alguna afección durante su curso, así como el grado de consanguineidad de los padres que como secuela produzca el trastorno de migración neuronal (21).
Conductas como la aplicación de técnicas para la determinación de la translucencia nucal, valoración anatómica fetal, evaluación de la superficie fetal (macizo facial, extremidades, genitales), evaluación del esqueleto fetal, medición de volumen (peso fetal, líquido amniótico y placenta), la confirmación de la presencia de anomalías, evaluación de la extensión y severidad de la malformación presente. Todo esto llevado a cabo con la ayuda de técnicas ecográficas, sin contar con la aplicación de técnicas invasivas como la biopsia de las vellosidades corionicas ayudaría a detectar cualquier alteración congénita.
Conflictos de interés
Los autores declaran no tener conflictos de interés.
Agradecimientos
Al Hospital Universitario Fernando Troconis, el cual nos suministró el permiso para acceder a la historia clínica de nuestro paciente, y principalmente a la madre de este quien nos mostró su apoyo y colaboración incondicional, y nos permitió el uso de la información clínico-patológica del niño.
Referencias
1. Correa J, Gómez J, Posada R. Fundamentos de pediatría Tomo V. 3a ed. Medellín Colombia: CIB; 2009.
2. Hernández M, Bolte L, Mesa T, Escobar R, Medallo C, Huete I. Lisencefalia y epilepsia en pediatría. Rev Chil Pediatr. 2007; 78 (6): 615-620. [ Links ]
3. Chang BS, Duzcan F, Kim S, Cinbis M, Aggarwal A, Apse KA, Ozdel O, Atmaca M, Zencir S, Bagci H,Walsh CA. 2007. The role of RELN in lissencephaly and neuropsychiatric disease. Am J Med Genet Part B 144B:58-63. [ Links ]
4. Valdivia Alvarez I, Abadal Borgues G. Epilepsia de difícil control en pediatría. Nuevas drogas antiepilépticas. Revcubpediatr. 2005 (77): 3-4. [ Links ]
5. Sadler TW. Langman Embriología médica con orientación clínica. 9a ed. Buenos Aires: Editorial médica panamericana; 2005.
6. Lambert de Rouvroit C, Goffinet A. Neuronal migration. Rev Mech Dev. 2001;(105): 47-56. [ Links ]
7. López Herrera J, García Ramírez R, Pérez Zárate M. Lisencefalia tipo I: síndrome de Miller-Dieker Informe de un caso. Rev Mex Pediatr. 1999; 66 (4): 157-160. [ Links ]
8. Bulakbasi N, Kocaoglu M, Üstünsoz B, Tayfun C, Somuncu I. MRI features of lissencephaly with cerebellar hypoplasia. CMIG Extra. Cases. 2004;(28):4-7. [ Links ]
9. Martino G, Martino R. Manual de epilepsia. 1a ed. Buenos Aires: Nobuko; 2007.
10. Nogales J, Donoso A, Verdugo R. Tratado de neurología clínica. 1a ed. Santiago de Chile: Editorial universitaria; 2005.
11. Pilz D. Miller-Dieker Syndrome. Orphanet J Rare Dis. 2003; 1(1): 1-3. [ Links ]
12. Cardoso C, Leventer R, Ward H, Toyo-oka K, Chung J, Gross A. Refinement of a 400-kb Critical Region Allows Genotypic Differentiation between Isolated Lissencephaly, Miller-Dieker Syndrome, and Other Phenotypes Secondary to Deletions of 17p13.3. Am J Hum Genet. 2003;(72): 918-930. [ Links ]
13. Sweeney K, Clark G, Prokscha A, Dobyns W, Eichele G. Lissencephaly associated mutations suggest a requirement for thePAFAH1B heterotrimeric complex in brain development. Rev Mech Dev. 2000;(92):263-271. [ Links ]
14. Cahana A, Reiner O. LIS1 and platelet-activating factor acetylhydrolase (Ib) catalytic subunits, expression in the mouse oocyte and zygote. FEBS Lett. 1999;(451): 99-102. [ Links ]
15. Abolila R, Alsawan R, AlrefaieM. Microcephalic osteodysplastic primordial dwarfism (MOPD) type I with lissencephaly and brain cyst. EJMHG (Egypt).2012;(13):363-365. [ Links ]
16. Leventer R, Guerrini R, Dobyns W. Malformations of cortical development and epilepsy. Dialogues ClinNeurosci. 2008; (10):47-62. [ Links ]
17. Bozzi Y, Casarosa S, Caleo M. Epilepsy as a neurodevelopmental disorder. Front Psychiatry. 2012;3:19. [ Links ]
18. Bart P. Disorders of neuronal migrations. Can J NeurolSci1987; (14):1-16. [ Links ]
19. Dobyns B, EstrattonR, Greenberg F. Syndromes and isolated lisencephaly sequence. Am J Genet 1984; (18): 509-26. [ Links ]
20. Alarcón Rodríguez R. Criptorquidias y xenoestrogenicidad por pesticidas en la provincia de Almería [tesis doctoral]. España: Editorial de la Universidad de Granada. Universidad de Granada, España; 2007.
21. Pascual-Castroviejo I, Velasquez R, Viaño J. Citomegalia congénita y malformaciones corticales y subcorticales. Neurología.2012;(27):336-42. [ Links ]
Rev. Méd. Risaralda 2014; 20 (2): 129-132
