










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkCT&F - Ciencia, Tecnología y Futuro
Print version ISSN 0122-5383On-line version ISSN 2382-4581
C.T.F Cienc. Tecnol. Futuro vol.3 no.5 Bucaramanga Jan./Dec. 2009
MOLECULAR AND MULTISCALE MODELING: REVIEW ON THE THEORIES AND APPLICATIONS IN CHEMICAL ENGINEERING
Giovanni Morales Medina* and Ramiro Martínez Rey
Escuela de Ingeniería Química, Universidad Industrial de Santander, Bucaramanga, Santander, Colombia , A.A. 678.
e-mail: gmorales@uis.edu.co e-mail: rmartine@uis.edu.co
(Received March 3, 2009; Accepted August 12, 2009)
*To whom correspondence may be addressed
ABSTRACT
We call molecular modeling to the application of suitable laws in the analysis of phenomena occurred at scales less than those accounted for by the macroscopic world. Such different scales (including micro-, meso- and macroscales), can be linked and integrated in order to improve understanding and predictions of complex physical chemistry phenomena, thus originating a global or multiscale analysis. A considerable amount of chemical engineering phenomena are complex due to the interrelation among these different realms of length and time. Multiscale modeling rises as an alternative for an outstanding mathematical and conceptual representation of such phenomena. This adequate representation may help to design and optimize chemical and petrochemical processes from a microscopic point of view. Herein we present a brief introduction to both molecular and multiscale modeling methods. We also comment and examine opportunities for applying the different levels of modeling to the analysis of industrial problems. The fundamental mathematical machinery of the molecular modelling theories is presented in order to motivate the study of these new engineering tools. Finally, we show a classification of different strategies for applying multilevel analysis, illustrating various examples of each methodology.
Keywords: multiscale modeling, molecular modeling, quantum chemistry, DFT, ab initio, molecular mechanics, QM/MM.
RESUMEN
El modelamiento molecular consiste en la aplicación de leyes apropiadas en el análisis de fenómenos que ocurren a niveles o escalas inferiores a la macroscópica. Visionariamente, estas escalas (incluyendo micro, meso y macro escalas), pueden ser acopladas en un modelo matemático global con el objetivo de mejorar el entendimiento de los fenómenos fisicoquímicos complejos. Muchos fenómenos a nivel industrial, sino todos, son también complejos y, por lo tanto, el análisis global aparece como una alternativa para la ingeniería química. La adecuada representación de las interacciones entre los diferentes niveles por medio del modelamiento multiescala puede ayudar, en un futuro próximo, en el diseño y la optimización de procesos químicos desde un punto de vista microscópico. En este artículo se presenta una breve introducción a las teorías del modelamiento molecular y multinivel. Así mismo, se discuten diferentes oportunidades para la aplicación de estas teorías en el análisis de procesos industriales. Una breve descripción matemática de los métodos moleculares es presentada con el fin de motivar el estudio de estas nuevas herramientas ingenieriles. Finalmente, las diferentes estrategias para aplicar el análisis multiescala son clasificadas e ilustradas con casos reportados recientemente en la literatura.
Palabras Clave: modelamiento molecular, modelamiento multiescala, química cuántica, DFT, ab initio, mecánica molecular, QM/MM.
RESUMEN
A modelação molecular consiste na aplicação de leis apropriadas na análise de fenˆmenos que ocorrem a níveis ou escalas inferiores à macroscópica. Visionariamente, estas escalas (incluindo micro, meso e macro escalas), podem ser acopladas em um modelo matemático global com o objetivo de melhorar o entendimento dos fenˆmenos físico-químicos complexos. Muitos fenˆmenos a nível industrial, senão todos, são também complexos e, portanto, a análise global aparece como uma alternativa para a engenharia química. A adequada representação das interações entre os diferentes níveis por meio da modelação multiescala pode ajudar, em um futuro próximo, no desenho e a otimização de processos químicos desde um ponto de vista microscópico. Neste artigo apresentase uma breve introdução às teorias da modelação molecular e multinível. Assim mesmo, discutem-se diferentes oportunidades para a aplicação destas teorias na análise de processos industriais. Uma breve descrição matemática dos métodos moleculares é apresentada com o fim de motivar o estudo destas novas ferramentas da engenharia. Finalmente, as diferentes estratégias para aplicar a análise multiescala são classificadas e ilustradas com casos reportados recentemente na literatura.
Palavras Chave: modelação molecular, modelação multiescala, química quântica, DFT, ab initio, mecânica molecular, QM/MM.
INTRODUCTION
Due to its complexity, modeling the real world becomes a challenge. Phenomena at different scales of size and time are often involved within systems, meanwhile, these are treated and processed in chemical and petrochemical plants. These size and time scales are related to different approaches in the movement and interaction of bodies that must be heeded for developing systematic procedures for the design and optimal operation of chemical plants; see the third paradigm for a modern chemical engineering (Charpentier, 2009). Conventionally, process engineering ranges its tools in the macroworld. However, necessities such as new characteristics of the products, and environmental restrictions, have motivated chemical engineers to think about body interactions at low levels or scales and include molecular-based models in the equations for describing a process. The study of the relationship between the scales and the inclusion of molecular-based models can provide the industry with new "tools" to design and operate processes effectively, in such a way that they might prove successful in today’s competitive commerce. Chemical engineering is entering a new era, characterized by unprecedented control over chemical reactions, as well as product molecular architecture, conformations and morphology (de Pablo, 2005). Experiments and processes are being interpreted and predicted through a multi-scale coupling (e.g. see Sengupta, 2003). The purpose of this review is to provide engineers with some key concepts about molecular modeling theories as well as to present some applicative examples of multiscale modeling in engineering phenomenon analysis.
ANALYSIS OF ENGINEERING PHENOMENA
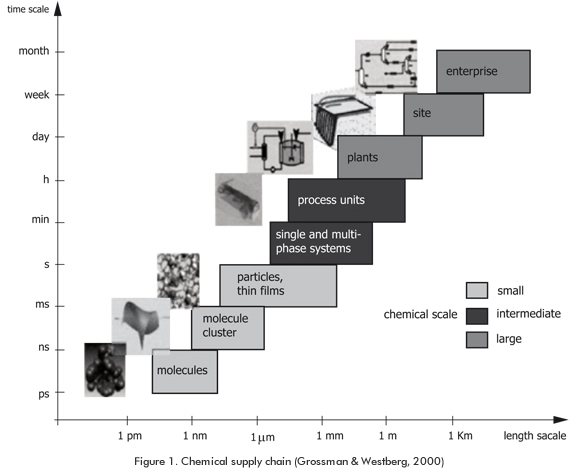
In spite of modern methods of mathematical analysis, conceptual representation of a real phenomenon (e.g., a chemical process) cannot completely encompass all the details of the process. Alternatively, the conceptual representation can be broken down into distinguishable subsystems which, when assembled into a whole, can simulate the process (Figure 1). These subsystems are classified according to their different body interactions, which depend on magnitudes of time and length. Such classification originates nanoscopic, atomistic, microscopic, mesoscopic, and macroscopic scales. Nanoscale is the scale that covers times less than nanoseconds and lengths less than 10 nm, being utilized for predicting molecular properties (e.g. polarizability and hydrogen bond), for which electronic distribution plays the major role (Westmoreland et al., 2002; Hung, Franzen, & Gubbins, 2004). As electronic structure is the main concern, quantum mechanical theory rules the behavior within this domain. Atomistic subsystem is enclosed in magnitudes of length and time greater than those in the nanoscale (i.e. 1nm - 0,1μm and 1ps - 1ns). Central issue in this level corresponds to evaluate interactions among large number of atoms and molecules and how such interactions influence the macroscopic properties of the subsystem. This scale uses force fields (classical mechanics) to describe the interaction between the molecules, and statistical mechanics to determine thermodynamic and transport properties. Therefore, the explicit influence of the electronic structure of atoms and molecules is lost in atomistic simulations (molecular mechanics). The atomistic simulates movements of the molecules either stochastically or deterministically. The simulation that is done in a stochastic manner receives the name of Monte Carlo and it is based on a probabilistic approach to the relative position of the atoms. On the other hand, simulations that describe the movement of molecules in a deterministic fashion are called molecular dynamics, and they are based on Newton’s laws of movement. Methods and theories involved in the two first scales of analysis are grouped in the so-called molecular modeling area.
Microscopic and mesoscopic scales cover from microns to millimeters and their time scales span from nanoseconds to milliseconds or even seconds. Phenomena encountered in these scales are currently out of reach of the atomistic simulation view. The central entities in the micro and mesoscales are groups of atoms or molecules ("beads of matter") which can interact through effective potentials (Hung et al., 2004). Highly complex topologies can be analyzed by using an effective potential such as the harmonic spring force. The main concern here is the simulation of phenomena found in systems constituted for several homogeneous matters, such as that encountered in ternary polymer melts (Posel, Lísal, & Brennan, 2009) as well as blood flow (Dzwinel, Yuen, & Boryczko, 2006); see other applications in Young (2001) and Maiti, Wescott & Goldbeck-Wood (2005). Models and theories used for analyzing the microscale are dissipative particle dyna-mics (DPD) (Hoogerbruge & Koelman, 1992; Koelman & Hoogerbruge, 1993), Ginzburg-Landau models (Glotzer, Stauffer, & Jan, 1994; Altevogt, Evers, Fraaije, Maurits, & van Vlimmeren, 1999) and Flory-Huggins theory (Fleer, Stuart, Scheutijens, Cosgrove & Vincent, 1993). Some of the methods applied in mesoscale modeling are lattice Boltzmann (Succi, 2001), stochastic rotation dynamics (Malevanets & Kapral, 1999), phase field models (Anderson, McFadden & Wheeler, 1998) and the Lowe-Andersen thermostat (Lowe, 1999).
The following domain in time and length is macroscale, which conceives matter as a continuous and properties of the systems as field quantities. Therefore, its mathematical formulation can efficiently handle phenomena at sizes greater than those treated in the preceding domains. In consequence, electronic and atomistic details as well as "bead" details are not explicitly included into the calculations. The mathematical modeling used in describing the operational regions into which equipments might operate has been mostly based on the use of the so-called MESH equations (mass balance, equilibrium equations, mole fraction summation and heat balance) and the momentum balance equation (Grossmann & Jackson, 2001). The greater disadvantage of the continuum macroscopic "tools" is the fact that they are, in their majority, explicitly independent on atomic structure relationship and might be unable to predict the physical-chemical consequences of new conditions, new compounds or new reactions in the systems thereof.
To achieve an in-depth control and simulation, the chemical engineering community must take into account the interactions in the nanoscopic, atomistic, microscopic and mesoscopic domains and, therefore, include molecular modeling as an important "tool" for the development and improvement of chemical supply chains (Figure 1). There are some areas where the molecular analysis is visualized as a common and strong "tool" for process engineering:
Design: There is an emphasis on reducing the costs in the developmental time by omitting some phases of the activity that have traditionally been considered vital in the conceptual design of chemical processes (e.g., omitting the pilot plant) (Doherty, 2001). Molecular analysis can contribute to improve procedures for process development by probing the feasibility of some steps in the design. One of the key initial steps at each level in the design procedure is to decide quickly what’s feasible and what’s not. Molecular modeling has an important role to play in supporting the different design activities by providing estimates of critical pieces of data that are missing in the early stages of process development.
Planning and operation: Molecular theories can help in the conception of manipulating chemical plants. Globally, a chemical plant must close mass and heat balances in order to model production. At this level, economical and environmental restrictions must be achieved in order to make chemical plants economically feasible. Decisions taken at planning-based level serve as targets for the process engineers to operate their plants. Different operational conditions must be considered and evaluated for achieving the target goals. However, as molecules are the common foundation for feedstock composition, different interactions inside streams may appear as operational conditions are tested (Figure 1). As a result of these interactions, molecules may behave in a completely different way and the process can generate streams with different properties than the wanted target properties. Accordingly, the hierarchy of decisions should be altered to consider the interactions at low scales into the planning models.
Simulation: Process that lead to microstructured products may include reaction and/or separation steps and, besides, structure-forming steps (e.g., producing crystals). The shape of a crystal produced by a process often has a major impact on product quality as well as processability. Estimating the shape of a crystal during the discovery and conceptual design phases of product and process development is of major value in many cases. Molecular theories may conduce to the estimation of interfacial phenomena in order to improve the shape prediction models (Doherty, 2001). Another example in which molecular modeling may help is the attainable region which is defined as a domain in the concentration space that can be achieved by using any system of steady-flow chemical reactors (Glasser, Hildebreant, & Crowe, 1987). For isothermal consecutive reactions, the attainable region depends on the value of the rate constants. Theoretical calculations can estimate the relative rates in good agreement with experimental data. For example, in the first theoretical study on ketene dimerization in solution, Morales, Martínez, & Ziegler (2008) reproduced the experimental yield for the symmetrical ketene dimer reported by Tenud, Weilenmann, & Dalwigk (1977).
Control: In systems such as nanobiological devices, micromachines, nanoelectronic devices, and protein microarrays and chips, the most important component is the control of events at nanoscopic and atomistic scales, in order to achieve desired final properties and product quality (Lidorikis, Bachlechner, Kalia, Nakano, & Vashishta, 2001; Braatz et al., 2006 and references therein). Simplified models or trial-and-error experimentation are used to design many of these devices. Moreover, a multiscale analysis detailing from nanoscopic to atomistic domains, can be utilized to design and control these sorts of processes. Recently, Braatz et al. (2006) illustrated how to use molecular modeling methods in order to achieve control in these processes.
Estimation of properties: When one or more of the chemicals in the process are dangerous or extremely expensive, physical properties are usually missing or difficult to measure. Theoretical calculations at molecular level provide a means of estimating missing properties including phase equilibrium. Moreover, molecular modeling can help to complement and resolve experimental measurements and discrepancies in regards to right molecular and bulk properties (Westmoreland et al., 2002; Sumathi & Green, 2002).
Application of theoretical procedures of high accuracy is usually hampered by the size of the system under study. However, molecular modeling can allow application of empirical methods on compounds of high molecular weight by giving property values for a set of base small molecules. For example, Morales & Martínez (2009), by using the CBS-Q multilevel method and the MVLR procedure, assessed missing group additivity values for predicting ketene polymer’s thermochemical properties.
Local group interaction parameters for empirical liquid-phase thermodynamic models, such as UNIQUAC and UNIFAC, can also be assessed by the use of theoretical calculations (see Klamt, 2005).
Researching: New developments in different areas such as materials (Westmoreland et al., 2002), catalysis (Woo, Margl, Deng, Cavallo, & Ziegler, 1999), polymers (Çagin et al., 2001), corrosion (Gómez et al., 2005), electronics (Lidorikis et al., 2001) and drug design (Drummond & Sumpter, 2007) can be guided by molecular modeling "tools". Developments in these areas can be firstly tested by using computational experimentation to obtain probes of feasibleness. These probes can justify experimentation in order to synthesize the compounds with the required properties.
The chemical engineering community has already started to use the "tools" of the molecular theories to investigate in some of the areas described above. There is no doubt that the molecular analysis is playing an increasingly important role in future process engineering research and practice (Charpentier, 2002). Efforts of the community have been channeled through AIChe in a series of international conferences called Foundations of Molecular Modeling and Simulation (FOMMS) in which industrial applications are collected to motivate the use of molecular theories. Likewise, the European symposium called ESCAPE includes topics targeting the same goal.
MOLECULAR MODELING METHODS
Molecular Modeling can be defined as the development and application of physical and chemical theories in the description of a phenomenon whose model, exclusively solved by computer, predicts the behavior of the phenomenon. This definition involves two principal issues in molecular modeling: the theories and the computer calculations. Development of these issues improves the quality of the predictions of the phenomenon. It is worth mentioning that the developments of computational techniques have revolutionized molecular modeling to the extent that most calculations could not be performed without the use of a computer (Van Speybrook, 2001). Later on, we will describe the theories enclosed in the modeling at nanoscale. Foundations about the simulation at atomistic and mesoscopic domains have been reviewed by other authors (Frenkel and Smit, 1996; Ungerer, Lacher, & Tavitian, 2006).
Quantum mechanical description of molecular systems
Quantum mechanics, QM, explicitly represents the electrons in a calculation, making it possible to derive properties that depend upon the electronic distribution (e.g. chemical reactions). Postulates and theorems of QM assert that microscopic systems are described by wave functions that completely characterize all of the properties of the system. In particular, there are QM operators corresponding to each physical observable that, when applied to the wave function, allow one to predict the probability of finding the system to exhibit a particular value or range of values for that observable (Cramer, 2002). The typical form of the Hamiltonian operator takes into account five contributions to the total energy of a system: the kinetic energy of the α nuclei and i electrons, the attraction of the electrons to the nuclei with atomic number Z, and the internuclear and interelectronic repulsions (Levine, 2001; Cramer, 2002),
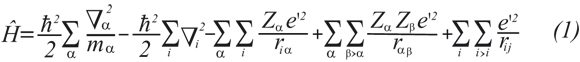
Then, the Schrödinger equation can be written in an operator manner according to Ĥψn(r)=Enψn(r).
The ψn term is the wave function that depends on both the r spatial coordinates and the n state of the system. En corresponds to the energy of the system in the n state. Exact solution of the Schrödinger equation is a very difficult task, mainly owing to the last two repulsion terms which have independent variables included into denominator. Internuclear repulsion term can be handled using the approximation of Born-Oppenheimer (static nuclei) that allows treating nuclei and electrons in a separate way (Levine, 2001; Cramer, 2002). According to this, an electronic Hamiltonian, Hele, for the system can be derived by canceling the first and fourth terms in equation 1. The interelectronic repulsion term, yet included into the Hele, can be treated from different approaches such as semiempirical and ab intio me-thods. Alternatively, density functional theory can treat the electronic system by solving the electronic density equation of Kohn-Sham instead of solving the Hele.
Semi-empirical (SE) molecular orbital methods are a computationally attractive alternative for ab initio methods, especially when medium and large molecules are subject of study. For treating the Hele easily, we can consider two kinds of approximation: in first place, Coulomb integrals are set to be zero in long distance interactions and, in the second place, interchange integrals are often replaced by analytical expressions with parameters, which are either experimentally or theoretically determined (Levine, 2001). Another distinctive approximation in SE methodologies is to take into account only valence electrons. SE methods are more expensive than the molecular mechanics method, but they allow breaking of bonds and take electronic effects explicitly into account, which molecular mechanics cannot. SE methods are appropriate, among others, for: a first step in a study of large and complex systems (see Klein et al. (2006)), and obtaining qualitative information about molecular properties (such as molecular orbitals, atomic charges and frequencies) (Foresman & Frisch, 1996). Important shortcomings of SE methods are low reliability (particularly for transition states and hydrogen bonding) and lack of reliable parameters for transition metals which are concerned in most catalytic phenomena (Foresman & Frisch, 1996; Westmoreland et al., 2002).
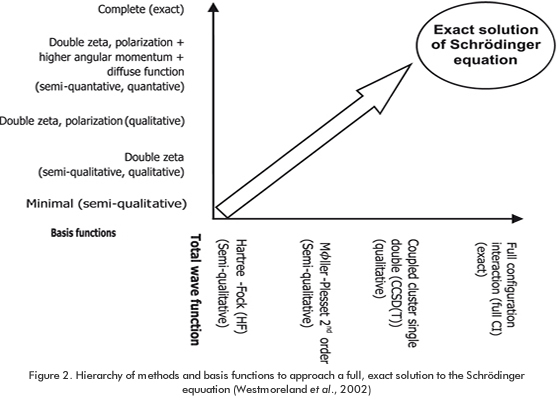
In regards to ab initio methods, we can state that all possible pairwise interelectronic repulsions (last term of equation1) in a molecular system can be treated in an ave-rage way (Hartree-Fock or HF approximation). The HF derived equation is solved for each electron of the system in an iterative method called self consistent field (SCF) (Levine, 2001; Cramer, 2002). The energies predicted with the HF approximation may not be accurate enough for chemical applications due to the neglect of the change of instantaneous position of an electron by the presence of other electrons (correlation) (Leach, 1996). However, HF allows tremendous progress by carrying out practical molecular orbital calculations. Thus, HF theory, in spite of its poorly significant fundamental conjectures, provides a very well defined stepping stone on the way to more sophisticated theories called ab initio (Latin for from the beginning’). The ab initio methods include the "electron correlation" by the use of Slater determinants (Levine, 2001). From a conceptual point of view, the Configuration Interaction method (CI) is probably the simplest method to improve HF results (Foresman, Head-Gordon, & Pople, 1992; Foresman & Frisch, 1996). CI includes excited states in the description of an electronic state and considers all possible combinations of the molecular orbitals. This full combination generates all po-ssible excitations and the full-CI wavefunction is obtained thereof. In practical terms, the excitations are truncated to a small fraction of excitations such as single (CIS) and double (CISD). An improvement of the CI method results can be done by also varying the coefficients of the basis functions. This approach is known as the multiconfiguration self-consistent field method (MCSSCF) (Leach, 1996). The excitations can also be incorporated by applying the Coupled Cluster theory (CC). The central tenet of the CC theory is that the full-CI wave functions can be described in terms of a truncated Taylor series expansion (Leach, 1996). A frequently used truncation considers only one-particle and two-particle excitation operators (CCSD). The CC method incorporates higher order excitations than the CI method. Figure 2 presents a hierarchy of the ab initio methods according to both the "basis set" and the "total wave function". The traditional ab initio approaches provide us a variety of techniques to obtain almost exact molecular properties to any desired accuracy (Van Speybroeck, 2002). The ab initio have been and will be used in industrial applications when the accuracy is needed or when less expensive alternative methods, such as SE or DFT methods, do not perform well (Westmoreland et al., 2002).
On the other hand, density functional theory (DFT) is a method that offers a useful tool to overcome the limitations of the computational demands of most of the ab initio methods. Based on the famous Hohenberg and Kohn theorems, DFT offers in principle an exact treatment of the electronic quantum problem (Van Speybroeck, 2002), and a sound basis for the development of computational strategies to obtain information about the energetics, structures, and properties of atoms and molecules at much lower costs than traditional ab initio techniques (Geerlings, De Proft, & Langenaeker, 2003). The basic variable of this theory is the one-electron density instead of the wavefunction. The first Hohenberg and Kohn’s theorem (Hohenberg & Kohn,1964) showed that all the ground-states properties, and therefore the wave function, were uniquely deterined by the charge density.
The Second Hohenberg and Kohn’s theorem provided a variational ansatz for obtaining ρ: searching for the ρ(r), minimizing the total electronic energy (Geerlings et al., 2003). With the DFT theorems, the problem of many-electron is stated as a three dimensional one-body density problem.
Although the power behind these theorems, it was only by the introduction of the Kohn-Sham equation (Kohn & Sham, 1965) that DFT started to be computationally workable. Kohn and Sham (1965) realized that the real system can be reduced to a non-interacting reference system (Koch & Holthausen, 2001). With this analogy, Kohn and Sham described the unknown functionals by the summation of the kinetic energy of the non-interacting reference system, the electron-nucleus interaction, the electron-electron Coulomb energy, and the exchange and correlation contributions, EXC[ρ];, that correct the results from the non-interacting system (Hohenberg & Kohn, 1964; Leach, 1996). The fundamental problem in a DFT calculation is to obtain the correct form for the EXC [ρ]; functional. Various a-pproximate exchange and correlation functionals have been proposed. The simplest are the local density based functionals (LDA), in which functionals depend only on ρ (Leach, 1996). The LDA approximation is not accurate enough for chemists for molecular modeling, though, and therefore the dependence on gradient of ρ must also be introduced into the EXC[ρ]; (Becke, 1985). This makes the calculated results acceptably enough at the chemist level. Some examples of the gradient-corrected functionals are Becke88-LYP and Becke88-Perdue86 as well as hybrid functionals, such as B3LYP, which mix the "exact HF" exchange with DFT in a somewhat empirical fraction in order to improve the energies further (Westmoreland et al., 2002).
Classical description of molecular systems: Molecular mechanics (MM)
The MM method was developed by Westheimer, Hendrickson, Wiberg, Allinger, Warshel and others (Burkert & Allinger, 1982) and it is applicable to organic and organometallic compounds as well as coordinative compounds of transition metals. MM withdraws the explicit influence of electrons and relies on considering that the energy of the system can be obtained by different interaction of punctual particles doted of mass and charge (atoms). These particles are joined by springs to allow representing molecules. Therefore, contributions to potential energy due to bonded atoms are obtained by bond stretching, angle bending and torsion contributions. MM models also permit non-bonded punctual particles to interact by considering electrostatic interactions and van der Waals interactions. Then, the energy of a system can be expressed as (Cramer, 2002; Leach, 1996),
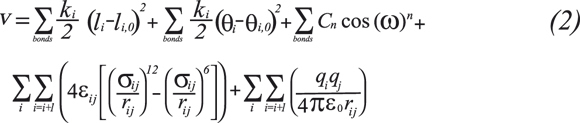
Where ki corresponds to force constant, li,0 and θ i,0 are the equilibrium values for bonds and angles, respectively. ω is the torsion angle. Set of terms for each of the energy contributions and parameter values in the potential energy equation is called force field (FF). The relative simple expression for the energy allows treating systems of thousands of atoms. Sophisticated FF may have additional terms, but invariably contain the components of Equation 2. The force fields classify atoms according to their hybridization and to the other atoms they are bonded to (Levine, 2001). The general procedure to obtain the values of the parameters in a force field consists in taking a training set of molecules with data collected from either experiments or theoretical ab initio calculations (Leach, 1996). Transferability is a key attribute of a FF, for it enables a set of parameters developed and tested on a relatively small number of cases to be applied to a much wider range of problems (Burkert & Allinger, 1982). MM methods are very fast and have proven their success to handle systems such as polypeptides or proteins with a huge number of atoms and stable conformations in both Monte Carlo and molecular dynamic simulations (McCammon & Harvey, 1987). In some cases FF can provide answers that are as accurate as even the highest level quantum calculations, in a fraction of computer time. Principal drawbacks of MM methods are related to poorness of general applicability due to their empirical input. They are additionally not suited to study bond-breaking and bond-forming reactions (Levine, 2001). Examples of FF available in literature are AMBER94 (Cornell, 1995) and CHARMM (MacKerell et al., 1998) for biomolecules, MMx for organic molecules, hydrocarbon and proteins (Allinger, Chen, & Lii, 1996), SHAPES for transition metal compounds (Allured, Kelly, & Landis, 1991) and PEF95SAC for carbohydrates (Fa-bricius, Engelsen, & Rasmussen, 1997).
Simulation at atomistic scale: Molecular simulation methods
A molecular simulation generally consists of a computer realization of a system in which actual molecular configurations are used to extract structural, thermodynamic and dynamic information (Frenkel & Smit, 2002). The term "configuration" denotes a set of cartesian coordinates (and momentum in the case of dynamic simulation) for all the atoms or molecules that constitute a system. Properties that can be obtained from molecular simulations include thermodynamic properties (such as equations of state, phase equilibrium, and critical constants), mechanical properties (such as stress-stretch relationships and elastic moduli), transport properties (such as viscosity, diffusion and thermal conductivity), and morphological information (such as location and shape of binding sites on a biomolecule and crystal structure).
Molecular simulation counts with two methods to generate system configurations as a direct consequence of the ergodic hypothesis (temporal average of a process parameter is equal to spatial average over the statistical ensemble). In molecular dynamics method (MD), the system is described by integrating its Newton’s equations of motion and, therefore, the parameters obtaining are time-averaged. The potential is modeled as a sum of interatomic potentials with a simple analytical form (molecular mechanics).
Initial positions for integration are chosen in a somewhat arbitrary manner due to the fact that simulation time is long enough for the equilibration of the system; on the other hand, initial velocities are established using the Maxwell Boltzmann distribution at the temperature of interest. Numerical integration of the equations of motion, along with the boundary conditions and any constraints on the systems, is done with the Verlet algorithm and its variants (Verlet, 1967; Allen & Tildesley, 1987). The solution of equation of motion simulates the microcanonical or NVE ensemble and, therefore, the energy (i.e. the sum of kinetic and potential energy) remains constant at any point over the phase-space trajectory. In order to perform a molecular dynamics calculation at imposed temperature (i.e. NVT ensemble), a thermostat method must be used to change the total energy of the system during the simulation, so that the Boltzman distribution of energies is fulfilled. In a thermostat method, the energy is changed by altering particle velocities at regular intervals (Nosé, 1984; Hoover, 1985). MD is especially suitable for studying the evolution of a non equilibrium structure or the dynamics of transport phenomena. From results of MD it is possible to calculate diffusion coefficients, thermal conductivity and viscosity.
On the other hand, in Monte Carlo methods (MC), spatial samples are generated under the restriction of a probability distribution function dictated by statistical mechanics (canonic ensemble have a probability which is proportional to exp(-E/KT)) and therefore parameters obtaining are ensemble-averaged. On the basis of these configurations, appropriate statistical averages are performed to derive fluid properties that can be compared with experimental measurements (Ungerer, Lachet, & Tavitian, 2006). Classes of movements in a MC simulation depend on the analyzed system. For instance, Gibbs ensemble Monte Carlo method (GEMC) for the simulation of liquid vapor equilibrium treats two separate microscopic regions (one for each phase) and performs cycles with the following random movements (Panagiotopoulos, 1987; Panagiotopoulos & Stapleton, 1989): displacements, volume changes, particle transfers and particle exchange. The key step in the GEMC method is the particle transfer which becomes as cumbersome as the increasing in length of chain of the treated molecules. This produce prohibitively low acceptance of transfer attempts. Particle transfer in GEMC can be improved by configurational-bias sampling techniques which are based on segment-by-segment insertions or removals of a multisegment molecule (de Pablo, Laso, Siepmann, & Suter, 1993; Frenkel & Smit, 1996). Several trial directions are attempted for every segment insertion, and a favorable growth direction is preferentially selected for the segment addition. Each segment growth or removal step generates a correction factor that is incorporated into the overall acceptance criterion in order to correct for the bias introduced by the non-random growth along preferential directions (Panagiotopoulos, 2001). The goal in the GEMC is to calculate the densities, compositions and pressures of the two coexisting phases at equilibrium without including the analysis of phenomena in their common interface.
All methods of molecular simulation have the common lack of appropriate intermolecular potential functions which is often quoted as the most important barrier to overpass for widely application to problems of industrial interest. The development of major number of force fields for chemical process conditions is required for properly representing the phase and reaction equilibria. Currently, the chemical engineering community count on useful force fields for a limited range of systems, including alkanes, al-kenes, perfluoroalkanes, CO2, H2O and other low molecular weight species (Westmoreland et al., 2002).
MULTISCALE ANALYSIS: LINKING AND INTEGRATION OF THE REALMS
An important challenge in modeling is how to link the different methods available to cover the whole range of length and time scales of interest (Hung et al., 2004). The typical objective of multiple analyses is to predict macroscopic behavior, such as selectivity, conversion, pollutant level, hot spots, etc. from first principles (Vlachos, Mhadeshwar, & Kaisare, 2006). An industrial example of integration of different approaches can be found in the oil additive field (Westmoreland et al., 2002). A broad range of factors determine performance and release of an antiwear chemical additive in motor oil. Each of these factors can be evaluated by different modeling approaches, such as QM calculations (thermal, oxidative and chemical stability of the additive), statistical mechanics (miscibility of the additive in the oil), finite-element methods (influence of the films appeared under extreme operational conditions on mechanical properties of the materials) and economic evaluation (prices and demands). Industrial problems like this require a multiscale analysis to develop solutions that meet the needs of industry. Linking and integration in multiscale modeling can be done, in general, in two fashions (Vlachos et al., 2006; Karakasidis & Charitidis, 2007); vide Ingram, Cameron y Hangos (2004) for an alternative classification. In the first one, named step-by-step procedure or hierarchical method, the integration is done in different sequential converging cycles and has the common characteristic that small level methods generate required input data for large level methods. Thus, input conditions of each subsystem in a step-by-step multilevel examination remain constant through each single modeling method. An example of step-by-step mode-ling can be analyzed in the paper of Albo, Broadbelt, y Snurr (2006) who studied mass transport and residence times of particles in nonostructured membranes used in catalysis. The study focused on new membranes fabricated by a combination of anodic aluminum oxidation (AAO) and atomic layer deposition (ALD) (Pellin et al., 2005). This route offers many possibilities to adjust and control the contact between reagents and catalytic sites on the walls and the selectivity toward the desired products can be improved thereof. The understanding of mass transport inside the pores can help to design the optimal pore size for a particular application. The study by Albo et al. (2006) addressed pores of up to 150 nm in diameter and up to 5 µm in length which made it necessary to use the diverse methods of modeling hierarchically:
1. Atomistic scale: Self-diffusivity of species inside the pores was analyzed through molecular dynamics (MD) simulations of the system. Alumina walls and the Lennard-Jones interaction between a molecule and the pore were represented by oxygen atoms and by the slit-pore potential, respectively. The results indicated that the surface diffusion disappeared as the temperature of the system was increased. Therefore, Knudsen diffusion was found to be the predominant mass-transport mechanism inside the pores under the typical conditions for selective catalytic oxidation.
2. Enlarged atomistic level (with the identification of the Knudsen regime): the scheme of the Dual Control Volume Grand Canonical Molecular Dynamic simulation at Knudsen diffusion regime was utilized to access scales of time and length longer than those at atomistic level. The values of the obtained transmission probability indicated that the particles enter the pore multiple times before reaching the opposite end of the pore, especially for larger values of the aspect ratio of pore length to pore diameter. This fact can affect contact between the catalyst and diffusing molecules and therefore, influence the performance of the reactor operation. The trends obtained in the preceding multilevel analysis can help to design and operate catalytic membrane reactors based on AAO/ALD nonostructured materials.
Another step-by-step analysis by Torres, Morales, Suarez, & Sánchez (2009) demonstrated that coupling different levels of modeling can achieve a good understanding of reactions and, therefore, reduce the complexity of the mathematical modeling. Methyl ether sulfonation process (MES) carried out in a falling film reactor (FFR) presents multiple challenges such as the description of the reaction coordinate as well as the prediction of the profiles for momentum, mass and heat transfers. Simulation of the FFR was done with the help of a hierarchical modeling as follows:
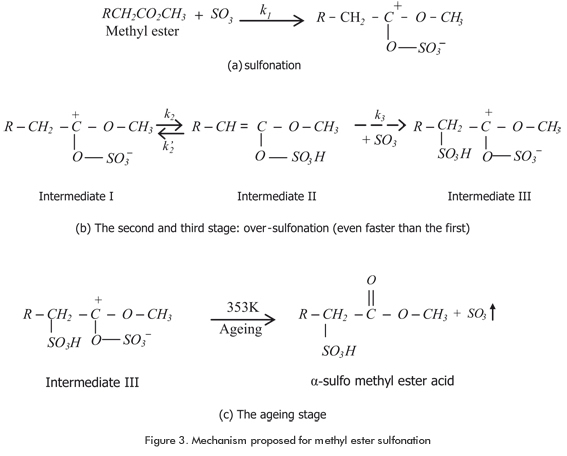
1. Nanoscale for MES reaction coordinate: Reaction steps for the MES were proposed as depicted in Figure 3. There is a slow third reaction stage where a SO3 group is liberated (on ageing). According to some researchers this SO3 group, just liberated, would be especially active and therefore capable of sulfonate directly another methyl ester molecule in a α position (Figure 3). This SO3 group may affect the mathematical expression for the kinetic of the process. The thermodynamic of the stable compounds for the MES process was analyzed at the B3LYP/6-31G(d) level. According to the results on the thermodynamic grounds, the intermediates produced in the sulfonation and over-sulfonation steps were found to have the same relative stability. This fact disregards the inclusion of these intermediates into the kinetic model and allows the use of a second order kinetic law for representing the reaction progress.
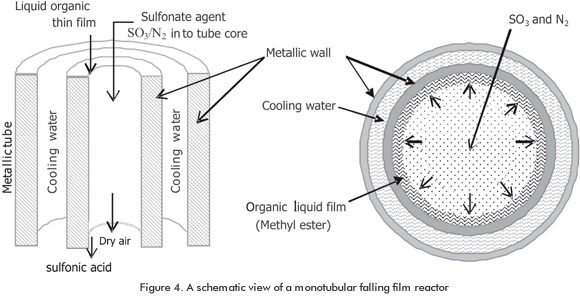
2. Microscale modeling for falling film reactor (Figure 4): The model proposed by Torres, Morales, Suarez, & Sánchez (2009) is appropriate for turbulent films and it considers effects of wavy film flow by using the eddy diffusivity parameter. The microscopic mass and energy balances are calculated by solving equations in partial derivatives for the liquid phase. Derived equations considered turbulent diffusivity for absorption with chemical reaction according to a second order kinetic law unveiled with the help of nanoscale modeling. This set of equations was numerically solved using Laasonen implicit forms for first and second order derivates. The results of this model reproduce the experimental data for fa-lling film reactors; thus, a fast conversion region at the top (gas phase control) and a slow conversion region at the bottom of the reactor (liquid phase control). The most important outlet data obtained by solving the mathematical model are the conversion of methyl ester, the density and viscosity of the sulfonic product. The proposed model can be suitable for use in design and operation of industrial falling film reactors even with petrochemical reactants.
There are many other examples in the literature in which multiscale modeling assists to analyze and identify important issues in different areas such as development of liquid-crystal based biosensors (Gupta, Skaife, Dubrovsky, & Abbott, 1998), methane partial oxidation and reforming (Mhadeshwar & Vlachos, 2005), self-assembled monolayers of thiophenes on electronic material surfaces (Haran, Goose, Clote, & Clancy, 2007), nanostructured materials and nanosystems (Scocchi, Posocco, Fermeglia, & Pricl, 2007; Fermeglia & Pricl, 2009a; Fermeglia & Pricl, 2009b) , modeling of reactors (Sengupta, 2003; Charpentier & McKenna, 2004; Majumber & Broadbelt, 2006; ), analysis of large biomole-cules (Huber, 2001), colloidal deposition (Kulkarni, Sureshkumar, & Biswas, 2005) and chemical product design (Morales-Rodríguez & Gani, 2009). In a simulation of the first homogeneous plug flow reactor for the production of ketene with a functionalized silica monolith, Martínez, Huff, & Barteau (2000) demonstrated that the theoretical value predicted for ketene’s heat of formation could reproduce the acetic acid conversion; see Martínez (2001) for more details.
In the second fashion of the multiscale analysis, named hybrid or concurrent method, the integration is done in the same main iteration cycle until achieving of appropriate convergence. The concurrent methods can incorporate detailed interactions and system information (such as spatiotemporal inhomogeneities on the surface) in a more computationally efficient manner than the single methods and can be divided into low-level and high-level hybrids. Developments on these methods have been concentrated on the low-level hybrids, i.e. in the integration between nanoscopic and atomistic domains, mainly. We called low-level hybrid methods those that integrate in the same iteration cycle QM methods and classic atomic scale methods. Accordingly, we can count three subclasses of low-level concurrent methods: temporal, spatial and spatiotemporal hybrids. Methods that are hybrid in a temporal sense merge electronic structure calculations (i.e., QM calculations) with molecular simulation methods. With this hybrid, the system is simulated at a finite temperature with an electronic structure method rather than an empirical force field. An example of this hybrid is the Car-Parrinello molecular dynamic method (Car & Parrinello, 1985), in which the atoms undergo motion described by classical dynamics in response to forces computed at the DFT level. This method and its various modifications have been very successful in material modeling and in calculations for heterogeneous and homogeneous catalysts in industrial applications (Westmoreland et al., 2002). The second class of low-level hybrids is related to divide the complex system spatially. In this sort, different methods are applied in different physical regions. Basically, this hybrid combines a more accurate (more expensive) method for the principal part of the system and a less accurate (less expensive) method for the rest of the system. Among this class of hybrid is the so-called QM/MM method (Warshel & Karplus, 1972) compound QM and MM by dividing the Hamiltonian into the form (Monard & Merz, 1999) H= HQM + HMM + HQM-MM, where HQM and HMM are the QM and MM Hamiltonians respectively. HQM-MM corresponds to the correction of the Hamiltonian by the interaction between the QM and the MM parts. This method allows bond breaking and forming processes of extended systems to be simulated in computationally tractable times (Woo et al., 1999). The QM part is the region that requires electronic distribution to be properly studied.
One of the first and main application areas has been the study of solvation and reactivity of small molecules, but other application areas include studies of surface reactivity, zeolites, crystal formation and study of reacti-vity in enzymes (Monard & Merz, 1999). A third class of low-level concurrent method, called spatiotemporal, can be formed when spatial and temporal hybrid methods are combined (Gao, 1992; Woo et al., 1999); i.e. the motion of the system is analyzed by molecular simulation and the spatial realm is treated by coupling QM and MM.
High-level concurrent methods resulting from the combination of both microscopic and macroscopic equations can improve the predictions of determining phenomena at detailed process conditions and therefore, they allow the appropriate change in operation to meet the require output of the systems. However, high-level hybrid methods that involve the combination between atomistic, mesoscopic and macroscopic scales are less common than the low-level hybrids that combine nanoscopic and atomistic levels. One of the reasons of that scarceness is the lack of developments in theories and procedures for proper coupling between these scales. Another reason is the computational time which can make the simulation prohibitive for most of the cases. The strategy for applying high-level concurrent methods is to spatially divide the system according to the scale of the phenomena to be coupled (Abraham, Broughton, & Bernstein, 1998). This strategy can be applied in semiconductor industry in which semiconductor devices, due to their size, show stress inhomogeneities caused by the high surface-to-volume ratio. Such inhomogeneities may affect the donor distribution by trapping donors in tensile stress regions (Lidorikis et al., 2001). Figure 5 illustrates the high-level hybrid methodo-logy for simulating stress distributions in a 25×25×1 nm Si (111) mesa, covered with a 25×25×5 nm Si3N4 film, on a 50×50×15 nm Si (111) substrate (Si/Si3N4 nanopixels) (Lidorikis et al., 2001). In this system, inclusion of atomically induced stresses at interfaces, where chemical bonding plays an important role, is enabled by applying MD simulation while most of the Si substrate is modeled by finite elements method (FE) as a continuum. MD atoms and FE nodes are merged in the handshake (HS) region. The elastic constants of the HS are matched with those of the "bulk" MD and FE systems for defining a total hybrid Hamiltonian (Lidorikis et al., 2001). Simulation with this high-level hybrid demonstrated the same behavior as that obtained with large scale MD simulation (Bachlechner, 1998), but at much less computational effort.
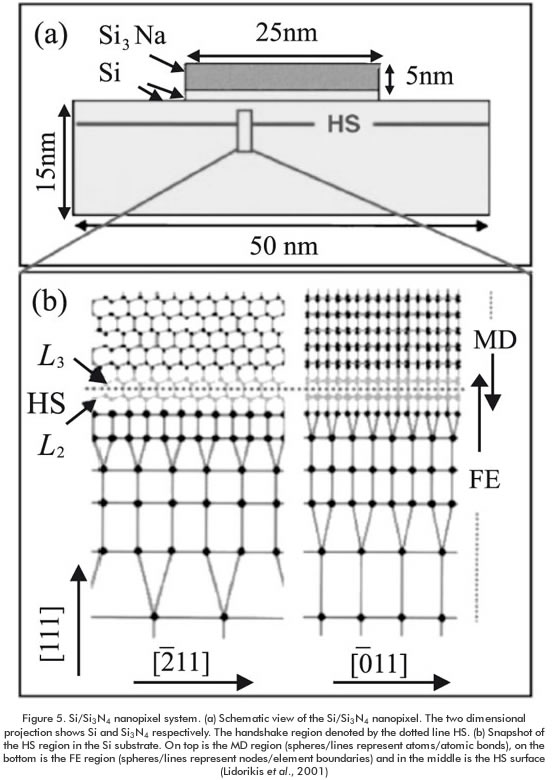
The two dimensional projection shows Si and Si3N4 respectively. The handshake region is denoted by the dotted line HS. (b) Snapshot of the HS region in the Si substrate. On top is the MD region (spheres/lines represent atoms/atomic bonds), on the bottom is the FE region (spheres/lines represent nodes/element boundaries) and in the middle is the HS surface (Lidorikis et al., 2001).
Another example of high-level hybrid method that couple macroscopic level with atomistic scale can be found in the work by Jensen, Hansen, Rodgers y Venkataramani (2001). In that work, different phenomena encountered in vapor deposition were analyzed in the same iteration cycle. The underlying physical and chemical process occurred at time and length magnitudes ranged from nanoscopic to macroscopic finite elements scale. Prediction of performance of deposited structure and the ultimate device requires understanding of how process conditions influence thin film synthesis on the atomic scale and therefore, multiscale analysis is needed to see this effect on macroscopic phenomena. FE that simulated the reactor macroscopically, and kinetic Monte Carlo (kMC) method, which models the evolution of the surface morphology during growth, were integrated through the flux of species to the surface (mesoscopic analysis). The kMC method uses the flux given by the FE method as input, and returns the computed flux which is in turn used by FE as a boundary condition at the substrate. The solutions of both problems are iterated until a consistent flux is determined.
The kMC method is embedded in the Newton iteration of the reactor scale FE model, which enables the linking through the surface flux boundary condition (Vlachos, 1997) and aids in convergence of the reactor scale model (Venkataramani, 2000). It is necessary to mention that neither chemical-physical transport at molecular level nor film morphology at micron scale can be studied by macroscopic conservation equations alone. Moreover, a step-by-step methodology can also fail in representing those ever-changing conditions (in CVD material is constantly exchanged between substrate and deposition chamber). This example demonstrates the needs of using multiscale analysis for understanding the complex processes. Other applications of high-level hybrid methods can be found in nanoelectronic devices (Cale, Bloomfield, Richards, Janse, & Gobbert, 2002; Windl, 2005) and in studying chemical reactors (Lin, Sureshkumar, & Kardos, 2001; Raimondeau & Vlachos, 2002). It is hoped that new developments on both theories and computational science facilitate the coupling between the domains, particularly in the case of chemical phenomena. As the systems to be analyzed become more complex and the accuracy required become higher, hybrid methods are expected to become more and more popular, in both academic and industrial applications, as the method codes become more readily available (Westmoreland et al., 2002) and the number of the specialized researchers increases.
CONCLUSIONS
- Modeling of chemical engineering phenomena based on different levels of the body interaction has started to contribute to the understanding and improving di-fferent steps of the chemical supply chain. The coupling among different scales of modeling arises as a useful tool for developing molecular-based algorithms which can represent detailed conditions of systems that different isolated models cannot take into account. Different coupling methods were analyzed and exemplified in this review to demonstrate the applicability of such methodology in different industrial topics. The examples demonstrated that molecular tools increase the understanding of the studied processes and can give suitable input data for mathematical models at microscopic and macroscopic scales. Future developments in theories, hardware and software can make multilevel coupling widely applicable, and the molecular modeling a common tool in the analysis of engineering problems.
ACKNOWLEDGEMENTS
The authors acknowledge the financial support of Universidad Industrial de Santander, UIS, and COLCIENCIAS (Instituto Colombiano Para el Avance de la Ciencia y la Tecnología Francisco José de Caldas).
REFERENCES
Abraham, F. F., Broughton, J. Q., Bernstein, N. & Kaxiras, E. (1998). Spaning the Length Scales in Dynamic Simulation. Comp. Phys., 12 (6), 538. [ Links ]
Albo, S. E., Broadbeltd, L. J. & Snurr, R. Q. (2006). Multiscale Modeling of Transport and Residence Times in Nanostructured Membranes.AIChe J., 52, 3679-3687. [ Links ]
Allen, M. P. & Tildesley, D. J. (1987). Computer simulation of liquids. Oxford Science Publications: New York. [ Links ]
Allinger, N. L., Chen, K. & Lii, J.-H. (1996). An Improved Force Field (MM4) for Saturated Hydrocarbons.J. Comput. Chem., 17: 642. [ Links ]
Altevogt, P., Evers, O. A., Fraaije, J. G. E. M., Maurits, N. M. & van Vlimmeren, A. C. (1999). The MesoDyn Project: Software for Mesoscale Chemical Engineering. THEOCHEM, 463: 139-143. [ Links ]
Allured, V. S., Kelly, C. & Landis, C. R. (1991). SHAPES Empirical Force Field: New Treatment of Angular Potentials and Its Application to Square-Planar Transition-Metal Complexes.J. Am. Chem. Soc., 113, 1. [ Links ]
Anderson, D., McFadden, G., Wheeler, A. (1998). Annu. Rev. Fluid Mech. 30: 139. [ Links ]
Bachlechner, M. E., Omeltchenko, A., Nakano, A., Kalia, R. K. and Vashishta, P., Ebbsjö, I., Madhukar, A. & Messina, P. (1998). Multimillion-Atom Molecular Dynamics Simulation of Atomic Level Stresses in Si(111)/Si3N4(0001) Nanopixels. Appl. Phys. Lett. 72: 1969-1971. [ Links ]
Becke, A. D. (1985). Local Exchange-Correlation Approximations and First-Row Molecular Dissociation Energies. Int. J. Quant. Chem. 27: 585-594. [ Links ]
Braatz, R. D., Alkire, R. C., Seebauer, E., Rusli, E., Gunawan, R., Drews, T. O., Li, X. & He, Y. (2006). Perspectives on the Design and Control of Multiscale Systems. J. Pro. Control, 16, 193. [ Links ]
Burkert, U. & Allinger, N. L. (1982). Molecular Mechanics. ACS monograph No. 177, American Chemical Society, Washington, D.C. [ Links ]
Çain, T., Wang, G., Martin, R., Zamanakos, G., Vaidehi, N., Mainz, D. T. & Goddard III, W. A. (2001). Multiscale Modeling and Simulation Methods with Application to Dendritic Polymers.Comp. Theor. Polymer Science, 11: 345-356. [ Links ]
Cale, T. S., Bloomfield, M. O., Richards, D. F., Jansen, K. E.,& Gobbert, M. K. (2002). Integrated multiscale process simulation". Comp. Mat. Science, 23: 3-14. [ Links ]
Car, R. & Parrinello, M. (1985). Unified Approach for Molecular Dynamics and Density-Functional Theory. Phys. Rev. Lett. 55: 2471-2474. [ Links ]
Charpentier, J. C. (2002). The Triplet "Molecular Processes-Product-Process" Engineering: The Future of Chemical Engineering?. Chem. Eng. Science, 57: 4667-4690. [ Links ]
Charpentier, J. C. (2009). Among the Trend for a Modern Chemical Engineering the Third Paradigm: the Time and Length Multiscale Approach as an Efficient Tool for Process Intensification and Product Design and Engineering. Chem. Eng. Res. Des., In press, doi: 10106/j.cher.2009.03.08. [ Links ]
Charpentier, J. C. and McKenna, T. F. (2004). Managing Complex Systems: Some Trends for the Future of Chemical and Process Engineering. Chem. Eng. Science, 59, 1617-1640. [ Links ]
Cornell, W. D. (1995). A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc., 117, 5179. [ Links ]
Cramer, C. J. (2002). Essentials of Computational Chemistry: Theories and Models. John Wiley & Sons, Ltd.: London. [ Links ]
De Pablo, J. J. (2005). Molecular and Multiscale Modeling in Chemical Engineering - Current View and Future Perspectives. AIChe J., 51, 2372. [ Links ]
De Pablo, J. J., Laso, M., Siepmann, J. I. & Suter, U. W. (1993). Continuum-Configurational-Bias Monte-Carlo Simulations of Long-Chain Alkanes. Mol. Phys., 80 (1), 55-63. [ Links ]
Doherty, M. F. (2001). Conceptual Design of Chemical Processes: Opportunities for Molecular Modeling. Foundations of Molecular Modeling and Simulation. AIChe Symposium Series 325, 97: 120-126. [ Links ]
Drummond, M. L. & Sumpter, B. G. (2007). Use of Drug Discovery Tools in Rational Organometallic CatalystDesign. Inorg. Chem., 46, 8613-8624. [ Links ]
Dzwinel, W., Yuen, D. A. & Boryczko, K. (2006). Bridging diverse physical scales with the discrete-particle paradigm in modeling colloidal dynamics with mesoscopic features.Chem. Eng. Sc., 61, 2169-2185. [ Links ]
Fabricius, J., Engelsen, S. B. & Rasmussen, K. (1997). The Consistent Force Field. 5. PEF95SAC: Optimized Potential Energy Function for Alcohols and Carbohydrates. J. Carbohydr. Chem., 16, 751-772. [ Links ]
Fermeglia, M. and Pricl, S. (2009a). Multiscale Molecular Modeling in nanostructured material desing and process system Engineering. Com. Chem. Eng., in Press, doi:10.1016/j.compchemeng. 2009.04.006. [ Links ]
Fermeglia, M. and Pricl, S. (2009b). Multiscale Molecular Modeling of Dispersion of Nanoarticles in Polymer Systems of Industrial Interest. IUTAM Symposium on Modelling Nanomaterials and Nanosystems, edited by Pyrz R. and Rauhe J. C., Springer-Verlag, Berlin, D, 1, 261-270. [ Links ]
Fleer, G. H., Stuart, M. A., Scheutijens, J. M., Cosgrove, T., Vincent, B. (1993). Polymers at Interfaces. Chapman and Hall: London. [ Links ]
Foresman, J. B., Head-Gordon, M. & Pople, J. A., Frisch, M. J. (1992). Toward a Systematic Molecular Orbital Theory for Excited States.J. Phys. Chem., 96, 135. [ Links ]
Foresman, J. B. & Frisch, Æ (1996). Exploring Chemistry with Electronic Structure Methods. 2nd edition, Gaussian, Inc., Pittsburg, PA. [ Links ]
Frenkel, D. & Smit, B. (1996). Understanding Molecular Simulation: From Algorithms to Applications. San Diego: Academic Press. [ Links ]
Gao, J. (1992). Absolute Free Energy of Solvation from Monte Carlo Simulations Using Combined Quantum and Molecular Mechanical Potentials. J. Phys. Chem. 96, 537-540. [ Links ]
Geerlings, P., De Proft, F. & Langenaeker, W. (2003). Conceptual Density Function Theory. Chem. Rev. 103, 1793 - 1873. [ Links ]
Glasser, D., Hildebrant, D. & Crowe,C. (1987). A Geometric Approach to Steady Flow Reactors: The Attainable Region and Optimization in Concentration Space. Ind. Eng. Chem. Res. 26, 1803. [ Links ]
Glotzer, S. C., Stauffer, D. & Jan, N. (1994). Phys. Rev. Lett., 72, 4109. [ Links ]
Gómez, B., Likhanova, N. V., Domínguez, M. A., Olivares, O., Hallen, J. M. & Martínez-Magadán, J. M. (2005). Theoretical Study of a New Group of Corrosion Inhibitors. J. Phys. Chem. A 109, 8950-8957. [ Links ]
Grossmann, I. E. & Westerberg, A. E. (2000). Research Challenges in Process Systems Engineering. AIChe J., 46 (9), 1700 -1703. [ Links ]
Grossmann, I. E. & Jackson, J. R. (2001). A Disjunctive Programming Approach for the Optimal Design of Reactive Distillation Columns. Comp. Chem. Eng., 25, 1661-1673. [ Links ]
Gupta, V. K., Skaife, J. J., Dubrovsky, T. B. & Abbott, N. L. (1998). Optical Amplification of Ligand-Receptor Binding Using Liquid Crystals. Science, 279, 2077. [ Links ]
Haran, M., Goose, J. E., Clote, N. P. & Clancy, P. (2007). Multiscale Modeling of Self-Assembled Monolayers of Thiophenes on Electronic Material Surfaces. Langmuir, 23, 4897-4909. [ Links ]
Hohenberg, P. & Kohn W. (1964). Inhomogeneous Electron. Gas. Phys. Rev. 136, B864 - B871. [ Links ]
Hoogerbruge, P. J.; Koelman, J. M. V. A. (1992). Europhys. Lett., 18, 155. [ Links ]
Hoover, W. G. (1985). Canonical Dynamics: Equilibrium Phase-Space Distributions. Phys. Rev. A, 31, 1695. [ Links ]
Huber, G. A. (2001). Multiscale Modeling of Large Biomolecules. Foundations of Molecular Modeling and Simulation. AIChe Symposium Series 325, 97: 54-60. [ Links ]
Hung, F. R., Franzen, S. & Gubbins, K. E. (2004). A Graduate Course on Multi-scale Modeling of Soft Matter, Chem. Eng. Ed., 38 (4), 242-249. [ Links ]
Ingram, G. D., Cameron, I. T. & Hangos, K. M. (2004). Classification and Analysis of Integrating Frameworks in Multiscale Modelling. Chem. Eng. Science 59, 2171 - 2187. [ Links ]
Jensen, K. F., Hansen, U., Rodgers, S. T. & Venkataramani, R. (2001). Bridging Length Scales in Simulations of Vapor Phase Deposition Processes. Foundations of Molecular Modeling and Simulation. AIChe Symposium Series 325, Vol. 97, 35-43. [ Links ]
Karakasidis, T. E. & Charitidis, C. A. (2007). Multiscale Modeling in Nanomaterials Science. Mat. Science Eng.C, 27, 1082-1089. [ Links ]
Klamt, A. (2005). Cosmo-RS. From Quantum Chemistry to Fluid Phase Thermodynamics and Drug Design. Elsevier. [ Links ]
Klein, M. T., Hou, G., Bertolacini, R. J. & Broadbelt, L. J., Kumar, A. (2006). Molecular Modeling in Heavy Hydrocarbon Conversions. CRC Press. Taylor & Francis Group. USA. [ Links ]
Koch, W. & Holthausen, M. C. (2001). A Chemist's Guide to Density Functional Theory. Second edition. Wiley-VCH Verlaf GmgH. [ Links ]
Koelman, J. M. V. A.; Hoogerbruge, P. J. (1993). Europhys. Lett., 21, 363. [ Links ]
Kohn, W. & Sham, L. (1965). Self-Consistent Equations Including Exchange and Correlation Effects. J. Phys. Rev., 140, A1133-A1138. [ Links ]
Kulkarni, P., Sureshkumar, R. & Biswas, P. (2005). Hierarchical Approach to Model Multilayer Colloidal Deposition in Porous Media. Environ. Sci. Technol., 39, 6361-6370. [ Links ]
Leach, A. (1996). Molecular Modeling: Principles and Applications. Longman, USA. [ Links ]
Levine, I. N. (2001). Química Cuántica. Segunda edición en Español. Prentice Hall, España. [ Links ]
Lidorikis, E., Bachlechner, M. E., Kalia, R. K., Nakano, A. & Vashishta, P. (2001). Coupling Length Scales for Multiscale Atomistics-Continuum Simulations: Atomistically Induced Stress Distributions in Si/Si3N4 Nanopixels. Phys. Rev. Lett., 87, 86104. [ Links ]
Lin, B., Sureshkumar, R. & Kardos, J. L. (2001). Electropolymerization of Pyrrole on PAN-Based Carbon Fibers: Experimental Observations and a Multiscale Modeling Approach. Chem. Eng. Science, 56, 6563-6575. [ Links ]
Lowe, C.P. (1999). Europhys. Lett. 37, 145. [ Links ]
MacKerell, A. D, Jr. , Bashford, D., Bellott, R. L., Dunbrack, R. L., Jr., Evanseck, J. D., Field, M. J., Fischer, S., Gao, J., Guo, H., Ha, S., Joseph-McCarthy, D., Kuchnir, L., Kuczera, K., Lau, F. T. K., Mattos, C., Michnick, S., Ngo, T., Nguyen, D. T., Prodhom, B., Reiher, W. E., III, Roux, B., Schlenkrich, M., Smith, J. C., Stote, R., Straub, J., Watanabe, M., Wiorkiewicz-Kuczera, J., Yin, D. & Karplus, M. (1998). All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B, 102, 3586-3616. [ Links ]
Maiti, A., Wescott, J. & Goldbeck-Wood, G (2005). Mesoscale modelling: recent developments and applications to nanocomposites, drug delivery and precipitation membranes. Int. J. Nanotechnology, 2, 198-214. [ Links ]
Majumber, D. & Broadbelt, L. J. (2006). A Multiscale Scheme for Modeling Catalytic Flow Reactors. AIChe J., 52, 4214-4228. [ Links ]
Malevanets, A. and Kapral, R. (1999). J. Chem. Phys. 110, 8605. [ Links ]
Martínez, R., Huff, M. C. & Barteau, M. A. (2000). Synthesis of Ketenes from Carboxylic Acids on Functionalized Silica Monoliths at Short Contact Times. Applied Catalysis A: General, 200, 79-88. [ Links ]
Martínez, R. (2001). Catalytic Synthesis of Ketenes on Silica Monoliths at Short Contact Times. A dissertation submitted in partial fulfillment of the requirements of Doctor of Philosophy in Chemical Engineering. University of Delaware. [ Links ]
McCammon, J. A. & Harvey, S. C. (1987). Dynamics of Proteins and Nucleic Acids. Cambridge University Press, New York. [ Links ]
Mhadeshwar, A. B. & Vlachos, D. G. (2005). Hierarchical Multiscale Mechanism Development for Methane Partial Oxidation and Reforming and for Thermal Decomposition of Oxygenates.Rh. J. Phys. Chem. B, 109, 16819-16835. [ Links ]
Monard, G. & Merz, K. Jr. (1999). Molecular Mechanical Methodologies Applied to Biomolecular Systems. Acc. Chem. Res., 32, 904-911. [ Links ]
Morales, G., Martinez, R. & Ziegler, T. A (2008). Theoretical Comparison of Ketene Dimerization in the Gas and Liquid Phase. J. Phys. Chem. A, 112, 3192-3200. [ Links ]
Morales, G. & Martínez, R. (2009). Thermochemical Properties and Contribution Groups for Ketene Dimers and Related Structures from Theoretical Calculations. J. Phys. Chem.A., 113, 8683-8703. [ Links ]
Morales-Rodríguez, R. & Gani, R. (2009). Multiscale Modeling Framework for Chemical Product-Process Design. Comput. Aided Chem. Eng. 26, 495-500. [ Links ]
Nosé, S. (1984). A Unified Formulation of the Constant Temperature Molecular Dynamics Methods. J. Chem. Phys., 81, 511. [ Links ]
Panagiotopoulos, A. Z. (1987). Direct Determination of Phase Coexistence Properties of Fluids by Monte Carlo Simulation in a New Ensemble.Mol. Phys., 61, 813. [ Links ]
Panagiotopoulos, A. Z. & Stapleton, M. R. (1989). The Gibbs Method for Molecular-based Computer Simulation of Phase Equilibria.Fluid Phase Equilibria, 53, 133-141. [ Links ]
Panagiotopoulos, A. Z. (2001). Force-Field Development for Simion of Condensed Phases. Foundations of Molecular ling and Simulation. AIChe Symposium Series 325, Vol. 97, 61-70. [ Links ]
Pellin, M. J., Stair, P. C., Xion, C., Elam, J. W., Birrell, J., Curtiss, L., George, S. M., Han, C. Y., Iton, L., Kung, M. & Wang, H. H. (2005). Mesoporous Catalytic Membranes: Synthetic Control of Pore Size and Wall Composition. Catal. Lett., 102, 127-130. [ Links ]
Posel, Z., Lísal, M. & Brennan, J.K. (2009). Interplay between microscopic and macroscopic phase separations in ternary polymer melts: Insight from mesoscale modeling. Fluid Phase Equilibria, 283, 38-48. [ Links ]
Raimondeau, S. & Vlachos, D. G. (2002). Recent developments on multiscale, hierarchical modeling of chemical reactors. Chem. Eng. J. 90, 3-23. [ Links ]
Scocchi, G., Posocco, P., Fermeglia, M. & Pricl, S. (2007). Polymer-Clay Nanocomposites: A Multiscale Molecular Modeling Approach. J. Phys. Chem. B, 111, 2143-2151. [ Links ]
Sengupta, D. (2003). Does the Ring Compound [(CH3)2-GaNH2];3 Form During MOVPE of Gallium Nitride? Investigations via Density Functional and Reaction Rate Theories. J. Phys. Chem. B, 107, 291-297. [ Links ]
Succi, S. (2001). The Lattice Boltzmann Equation for Fluid Dynamics and Beyond, Clarendon Press, Oxford. [ Links ]
Sumathi, R. & Green, W. H. Jr. (2002). Thermodynamic Properties of Ketenes: Group Additivity Values from Quantum Chemical Calculations. J. Phys. Chem.A, 106, 7937. [ Links ]
Tenud, L., Weilenmann, M., Dallwigk, E. (1977). 1,3-Cyclobutanodionderivate aus Keten. Helv. Chim. Acta, 60, 975-977. [ Links ]
Torres, J. A., Morales, G., Suarez, O. Y. & Sánchez, F. J. (2009). Mathematical Model of a Falling Film Reactor for Methyl Ester Sulfonation. Chemical Product and Process Modeling, 4 (5), Article 12. [ Links ]
Ungerer, P., Lachet, V. & Tavitian, B. (2006). Applications of Molecular Simulation in Oil and Gas Production and Processing. Oil & Gas Science and Technology - Revue de I'IFP 61, 3, 387. [ Links ]
Valchos, D. G. (1997). Multiscale Integration Hybrid Algorithms for Homogeneous-Heterogeneous Reactors. AIChe J., 43, 3031-3041. [ Links ]
Vlachos, D. G., Mhadeshwar, A. B. & Kaisare, N.S. (2006). Hierarchical Multiscale Model-Based Design of Experiments, Catalysts, and Reactors for Fuel Processing. Comp. Chem. Eng., 30, 1712-1724. [ Links ]
Van Speybrook, V. (2001). Ab Initio and Dynamic Molecular Methods: A Useful Tool in the Study of Chemical Reactions. Proefschrift Ingediend tot het Behalen Van de Graad Van Doctor in de Toegepaste. Faculteit Toegepaste Wetenschappen, Belgium. [ Links ]
Venkataramani, R. (2000). Multiscale Models of the Metaloganic Vapor Phase Epitaxy Process. A Dissertation Submitted as a Partial Requirement for the Degree of Doctor. Massachusetts Institute of Technology, Dept. of Chemical Engineering. [ Links ]
Verlet, L. (1967). Computer Experiments on Classical Fluids. I. Thermodynamic Properties of Lennard-Jones Molecules. Phys. Rev., 159, 98-103. [ Links ]
Warshel, A. & Karplus, M. (1972). Calculation of Ground State and Excited State Potential Surfaces of Conjugated Molecules. I. Formulation and Parametrization. J. Am. Chem. Soc., 94, 5612. [ Links ]
Westmoreland, P. R., Kollman, P. A., Chaka, A. M., Cummings, P. T., Morokuma, K., Neurock, M., Stechel, E. B. & Vashishta, P. (2002). WTEC panel report on applications of molecular and materials modeling. International Technology Research Institute. World Technology Division. [ Links ]
Windl, W. (2005). Ab Initio Assisted Process Modeling for Si-Based Nanoelectronic Devices. Mat. Science Eng. B, 124-125, 62-71. [ Links ]
Woo, T. K., Margl, P. M., Deng, L., Cavallo, L. & Ziegler, T. (1999). Towards More Realistic Computational Modeling of Homogenous Catalysis by Density Functional Theory: Combine QM/MM and Ab Initio Molecular Dynamics. Catalysis Today, 50, 479-500. [ Links ]
Young, D. C. (2001). Computational Chemistry. John Wiley & Sons, Inc. [ Links ]

