











 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
Permalink1. INTRODUCTION
One of the most critical parameters for the quality evaluation of natural gas is the presence of heavy hydrocarbons condensates, which lead to the detriment to the integrity of the pipelines. These hydrocarbons might condensate in a broad range of operating conditions. Hence, these should be removed before their compression, using efficient separation methods. For such reason, the gas is typically expanded, decreasing its pressure down to 35 bars inducing the condensation and removal of the produced liquid, according to the cricondentherm condition. Afterwards, the gas is re-compressed to be injected into the transport networks, a process that increases the cost due to the high-energy consumption (ca. 12% of the transportation cost) [1]. The implementation of a high pressure-phase separation process is a viable alternative to avoid the loss of energy through the expansion-recompression method [2]. However, during high-pressure gases separation, the formed condensate drops have a small diameter distribution of the order of micrometers, due to surface tension decrease as the pressure increases [2]; complicating the separation. Experiments conducted by Havelka et al [3] and referenced by Brigadeau [2], illustrates the disintegration of jets of n-decane within a broad range of pressures. They found that at lower pressures the disintegration follows a regular Rayleigh break-up (drop formation whose dimensions are significantly larger than the jet diameter ([4]), while at higher pressures the jet changes into a spray. These small drops lack inertia make them difficult to separate from the gas flow.
According to the previously mentioned, the design of high-pressure liquid-gas separators requires the analysis of the molecular interactions that lead to the formation of the nucleus and the growth of droplets. [5],[6]. Measurements of these interactions might take to an estimate of the more trusted drop sizes distribution for the condensation process, than the empiric values currently used for the design of separators. In the last 70 years, efforts have been intensified to provide different nucleation models and new experimental measurement techniques to understand the phase change phenomenon (gas to liquid) in pure and multicomponent systems. Within the current models for multicomponent mixing, there is the Classical Nucleation Theory (CNT), the Semi-Phenomenological Theory (SPT), the Functional Density Theory (FDT) and the Molecular Simulation (MS).
According to Merikanto [7], the CNT has been developed with theory and experimental contributions of [16]-[19] and the modifications of Sir William Thomson (Lord Kelvin), [13]-[15].
This theory has as the central assumption that the formed nuclei are spherical and that the physical properties are the result of macroscopic contributions of the evaluating fluid. On the other hand, the SPT is a branch led by Kalikmanov [16]-[19], that began with the studies of Fisher [20] and the contributions of Dillmann [21], and Ford, et al [22]. These models are based on suppositions similar to the CNT for multicomponent mixes, but it uses estimations of surface tensions obtained from the statistical thermodynamics [20]. The starting point of the TDF is evidenced in the research carried out by Cahn [23] upon free energy in the non-uniformed systems applied later by Oxtoby [24]-[25], Napari [26], and Talanquer [27]. The model considers the drop suspended in a saturated vapor as a non-homogeneous fluid with a variable density profile according to its distance to the center. Lastly, the MS could use two technics to simulate the nucleation phenomenon: The Monte Carlo method (MC) and the Molecular Dynamic (DM). This branch with several applications in the multicomponent mixes but barely used for the analysis of the generation of condensates in natural gas (reduced to nonane methane mixes) has been studied by authors such as Frenkel [28]-[29], Chen [30]-[32], Romero [33], Allen [34], Salonen [35] and Toxvaerd [36] amongst others.
All experimental results, as well as the predictions models, show errors between two and three magnitude orders as described by Fransen [37] and Wedekind [38]. These are due to factors such as the uncertainty of the nucleation velocity measurements and the lack of knowledge of the properties of the fluids in the scale of interest. Besides, to the best of our knowledge, conducted studies have been focussed upon water condensation, heptane and binary mixes such as methane/nonane (the more similar to natural gas). In the case of water, [39]-[41] reported that nucleation velocity predicted by the Classical Nucleation Theory (CNT) and CNT with empiric corrections differ in four orders of magnitude respect to experimental values for nucleation of steam. The same comparison, but using predictions obtained by la Semi-Phenomenological Theory, showed differences of three orders of magnitude. The sub-estimation observed in the predictions of the CNT might be due to the capillarity approximation involved in these models [40]. On the other hand, a study carried out by Braun [42] showed that predictions of nucleation velocity obtained by molecular simulation and Semi-Phenomenological Theory (SPT) are similar.
For the case of multicomponent systems, [18] compared the predictions of the CNT and SPT against experimental results reported by Luijten [5], Peeters, [43], and Labetski et al [41]. Kalikmanov found that values of nucleation velocity predicted by the CNT and SPT are lower than those obtained experimentally and that this difference increases with increasing pressure. For the case of molecular simulation, [30] conducted different comparisons of the experimental results of condensation of pure components (heptane, pentane and other heavy) against molecular simulation using the Monte Carlo method. He found an acceptable agreement, due to the complexity of the molecules, with a sub estimation of up to two orders of magnitude concerning to the experimental results. On the other hand, Labetski [44] carried out studies of binary mixes and ternary (n-nonane/methane, methane/nonane, methane/ propane/nonane) at pressures close to 40 bar with the adequate correspondence between experimental results and results obtained by molecular simulation.
Alternatively, the application for molecular simulation, especially using the Monte Carlo Method [30] may lead to the acquisition of an estimated for the droplet nucleus diameter, according to the processes at the nanoscopic level. Different jobs where the molecular simulation has been applied, have reported results that lead to understand and validate the process of nucleation in gaseous systems with components which are susceptible to condensation. [18];[37];[31];[35].
Nevertheless, it is important to highlight that the nucleation systems analysed through the Monte Carlo simulation have been limited to mixes with a maximum of three components of the type alkane; this differs from the typical composition of the natural gas, having between 6 and 10 components. According to the above, this work was focussed upon the analysis of the nucleation process at different pressures for a mixture of gases of composition similar to that of natural gas (up to six components of the alkane type) through the Monte Carlo simulation. Additionally, the growth of the drop through a phenomenological model was evaluated. Results included in this document are a contribution to the understanding of the separation gas-liquid process within the hydrocarbons mix, adding to the construction of fundamental procedures toward the design and adequate operation of condensate separation equipment.
2. THEORETICAL FRAMEWORK
The process of condensate drops formation involves three consecutive stages known as supersaturation, nucleation and droplets growth. The supersaturation occurs in the gas phase when the concentration of a component at the temperature and pressure of the system, at a given position, exceeds the respective concentration of the vapor-liquid equilibrium ([18). The driving force for the condensation of molecules of components in the gas phase corresponds to changes in the chemical potential [45]. Mathematically, the supersaturation for a system of one or more components can be determined according to:

Where P v , P sat (T),T y fe correspond respectively to the vapor pressure of the assessed component, the saturation pressure of the pure component, the T temperature and the correction coefficient of the ideal gas system [18]. Therefore, if S < 1 there is no formation of condensate drops; otherwise, if S >1 (supersaturated system) there is a high probability of nucleation-agglomeration appearance with enough quantity of molecules that causes the formation of an incipient drop or cluster (n *). The Equation 1 was used for the assessment of S in this study and was measured for all components of the assessed mixtures as proposed by Luijten [5] and Peeters [46]. The energetic barrier corresponding to the required free energy for nucleation is given by Kalikmanov [17].

Where G(n) is the formation free energy of clusters which is a function of the number of the molecules of the cluster (n) and go is the energy for each molecule in the vapor phase. Nevertheless, ΔW could be read like the work of formation of a cluster in the system at constant temperature and pressure [47]. The value of n where the value of ΔG required for the phase change is reached corresponds to the critical cluster n *. This condition is considered as a transition state, as the formed cluster could continue to grow -a reduction of free energy- or could experiment disassociation (n <n* ). [28] Defined the probability function that describe the distribution of the critical cluster as P(n)=(N n)/N, where (N n) corresponds to the average number of the nucleus with n molecules, and N is the total number of molecules in the system. The value of (N n) is established in terms of the free energy of Gibbs of the cluster, as:

This value matches the number of average nuclei detected in the different configurations obtained during a simulation process. Starting from Equation 3, the function of the distribution of the probability of nucleus formation can be defined as:

Once the nucleus has been formed, it starts adding molecules until reaching equilibrium; i.e., the nucleus moves from a size of the order of nanometers to micrometers [48]-[49]. According to Lebon, Jou [50], there are two limiting regimes in which the growth of the drop could take place, depending upon the value of the Knudsen number (K n), defined as the ratio of the molecular mean free path length of a vapor respect to the drop diameter. If K n < 0.1, there is little movement of molecules; this is a typical situation when the gas pressure is higher, and the growth is dominated by the diffusion of vapor molecules. If K n > 0.1, there is more space for the molecules free movement as is the case of the initial stage of the growing process; this is controlled by the crashing of individual molecules. Therefore, the K n value allows the definition of the boundary conditions in the mass and energy balances for the study and prediction of the behavior during growth. The analysis of the growth stage and the estimated final of the drop size in equilibrium can be conducted according to Young proposed models [51] and Gyarmathy [52] based upon mass and energy balances, depending on the relevant regime.
3. METHODOLOGY
NUCLEATION SIMULATION.
The nucleation of the heavy components of natural gas was conducted using the Monte Carlo method, codified in MATLAB®. The isothermal-isobaric ensemble (NPT) was considered as reported in the works of Wolde [28], Chen et al. [32], Fransen [37], and Shen [53]. The natural gas composition was simulated through a mixture of normal alkanes up to hexane including nonane. The total energy of the system was estimated using the potential of Lennard-Jones and intramolecular interactions by tension, flexion, and torsion. The joined atom method, called TraPPE-UA [54] was applied to consider the methyl and methylene groups as a particle with simple interaction.
The Monte Carlo simulation implemented the standard movements for NPT (translation, change of volume) and included the configurational biased methodology (CBMC) to improve the simulation efficiency of the flexible molecules. The cutting distance for the intermolecular and intramolecular interactions was set at 2.5 times the reference atomic diameter (9.3 A) [55].
The initial stage of the Monte Carlo simulations led to 104 cycles to acquire equilibrium. This number of cycles was defined from simulations carried out using the NPT ensemble [34], the compositions provided in Table 1, in a range of pressures and temperatures between 20 and 40 bar, and 240 and 265 K respectively. In this case, it was monitoring the variation of density as an indicator parameter of the convergence of the simulation. For this work, it was agreed that the convergence is reached when the standard deviation of the last 500 cycles is less than 2%.
Table 1 Composition for evaluated mixes.
| Component | Units | Heptane | Methane/Nonane | Natural Gas |
| Methane | [mol/mol] | - | 0.99 | 0.81 |
| Ethane | [mol/mol] | - | - | 0.025 |
| Propane | [mol/mol] | - | - | 0.01 |
| n-Butane | [mol/mol] | - | - | 0.01 |
| n-Pentane | [mol/mol] | - | - | 0,01 |
| n-Hexane | [mol/mol] | - | - | 0.01 |
| n-Heptane | [mol/mol] | 1.0 | - | - |
| n-Nonane | [mol/mol] | - | 0.01 | - |
| Nitrogen | [mol/mol] | - | - | 0.05 |
| Carbon Dioxide | [mol/mol] | - | - | 0.075 |
| Systems total pressure | kPa reduced | 6.5 | 2500 to 4000 0.03 to 0.1 | 2200 to 4000 0.1 |
| Temperature of nucleation/growth | K reduced | 245 1.6 | 240 to 265 1.6-1.8 | 240 1.6 |
Each cycle included 25 Monte Carlo movements randomly executed and distributed as follows: ten (10) translations, five (7) volume changes and five (8) re-growing of alkane molecules selected randomly [54]. The same number of cycles as previously stated was specified for the production stage hence different properties were averaged including the number and size of the formed nucleus.
It is essential to establish a consistent algorithm for the molecular nucleation simulation that allows identifying the existence of clusters. There is no current method of cluster detection applicable to all systems, but there are several alternatives. One of them is the geometric criterion [56] that establishing a dimensional restriction (minimum distance between particles) on the molecular locations. Other two are the criteria of energetic restriction [32] (maximum energy between particles) and the tWF [28]; this last consisting of a combination of geometric restriction with a minimum number of particles in the cluster. The tWF criterion was selected due to the implementation easiness and due to the fact that some authors who applied it [28];[57] obtaining results with the acceptable correspondence during the comparisons. For its use, a maximum distance between neighbouring molecules of 5.5 A was defined and a minimum number of 5 neighbours. The model proposed by Allen [34] was used as a reference for the development of the algorithm of cluster detection, modified for its application to formed molecules by pseudoatomic chains.
On the other hand, the calculation of the barrier of energy in function of the size of the cluster was made using the so-called umbrella sampling [58]-[60],[55,32,53,28], with the use of the potential harmonic bias [61,62,28], accordingly,

Where n is the size of the detected nucleus and nref, is the size of the referenced nucleus related to the criteria of the tWF cluster (5 molecules). The value of the strength constant depends upon the own characteristics of the simulated system. Wolde [28] recommends values between 0.01 and 0.1.
The umbrella integration method proposed by Stecher [63] and Kàstner [60] was used to perform the analysis of the simulation results using the umbrella method. This method estimates the probability of the drop formation (with a weighted average of the individual probabilities) obtained in each window resulting from the simulation, following the expression:
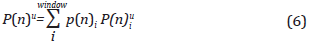
Where p(n)¡ is a weighted weight about the number of cycles used for each window. From P(n)u it is possible to obtain the Gibbs free energy barrier [30] and accordingly the critical cluster size.
SIMULATIONS OF DROPLET GROWTH.
The droplets growth was simulated according to the model proposed by Young [51], which represents a mathematical description of the mechanism of the gaining of molecules of a drop of liquid suspended in a gas, from equations of conservation of mass and energy. The model was used under such conditions that the Knudsen number is less than 0.1 and it considers a transition layer between the liquid and gas phases, so called the Knudsen boundary layer. The transition layer has a thickness estimated of the order of magnitude of the mean free path of molecules, so that collision between molecules are neglected, allowing to use the kinetic gases theory for the calculations of mass and energy flow. The mathematic model includes the mass and energy balances per region, and an equation to relate the density of the phases liquid and gas to the transition layer, resulting in a system of seven equations which once solved allows to obtain the fields of temperature and concentration, and the droplet diameter.
It is important to highlight that the Knudsen number was calculated as the ratio between the free mean path of the molecules (λ) to the droplet diameter (dp). The mean free path was calculated with the equation:

Where the n g is the molar density of the gas obtained from the gas density, and dg is the collision diameter, taken as the Lennard Jones diameter of the gas and estimated from the viscosity.
The simulation was executed taking into account the incoming gas composition data, pressure, temperature and the cluster size. The result is the droplet diameter, which can be used as input data for the design of gas separation systems in the natural gas industry.
4 RESULTS AND ANALYSIS
Simulations for the prediction of the size of the drop of condensate mixtures of alkanes similar to natural gas were conducted using compositions as per Table 1.
The system called "Heptane" was used to make comparisons with experimental nucleation results as described by Chen et al. [32]. The mixture "Methane/Nonene" was used to make comparisons of nucleation and the drop growth against the ones reported by Looijmans et )al., [64] and Luijten [5]. The speed of nucleation and the drop's size were estimated for a "Natural Gas' whose composition corresponds to that of Table 1.
The execution of the NPT ensemble cycles production and the umbrella sampling, to the operation conditions of 4 MPa and 240 K, produced nucleus with molecules number between 5 and 70, with calculated averaged diameters from 0.1 to 2.0 nm.
CALCULATION OF GIBBS FREE ENERGY.
a. Case "Heptane".
The simulations for the production step were carried out applying the umbrella method sampling with a force constant k = 0.01 [37]. The number of windows used was 6, each one with 10000 cycles and nucleus sizes between 5 and 150 (n ref) molecules; results indicated that the major nuclei size was that formed by 68 molecules. On the other hand, Figure 1a shows the dimensionless Gibbs energy (βΔG) found versus the number of heptane molecules conforming the critical nucleus. According to this Figure, the maximum energetic barrier value corresponds to 65 for a nucleus conformed by 52 heptane molecules. The trends show in Figure 1a are in agree with those reported by Chen et al. [32].
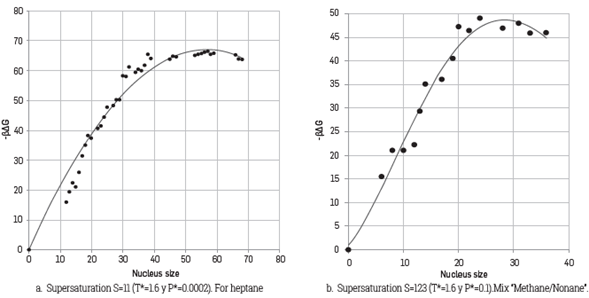
Figure 1 Energy barrier according to the cluster’s size. Heptane and “Methane/Nonane” (the continuous line represents averaged trends and circles represent the results from the simulation).
b. Case "Methane/Nonane".
For this case, 40000 molecules were used (with 60 nonane molecules) setting a pressure of 4 MPa and temperatures between 240 K and 265 K; this range of temperatures allowed stablishing the values of supersaturation, S, between 25 and 123. The production stage used the umbrella sampling method with a constant force k = 0.05, accounting for nucleus size values between 5 and 50. The larger size of the obtained nucleus during simulations was the one formed by 46 molecules. The variation of the dimensionless energy barrier is shown in Figure 1b, where it is possible to appreciate that the critical cluster is conformed by 26 molecules, 16 of which are of methane and 10 of nonane.
c. Case Natural Gas.
There are no results allowing comparing the nucleation for the case of the natural gas mix, but there are results for the droplets growth at 22 bar. Simulation results for the Gibbs free energy for the detection of the critical cluster at 22 bar and 40 bar are shown in Figure 2. For the case of simulation at 22 bar, a critical cluster was found, conformed by 22 molecules (3 hexane, 4 pentane, 2 butane, 3 propane and 10 methane); for 40 bar the critical nucleus reported 41 molecules (6 hexane, 4 pentane, 1 propane, 5 ethane and 25 methane).
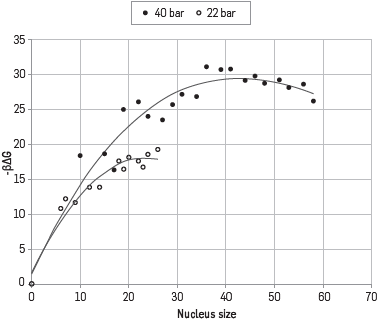
NUCLEATION VELOCITY COMPARISON.
The used parameter for comparison between experimental results and theory models is the velocity of nucleation J, that can be estimated through the given TNC equation, and simplified by Shen [53]:
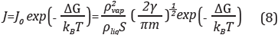
Where J 0 is a factor mainly depending upon temperature; this is calculated as the inversed value of supersaturation (1/S). Likewise, p vap and p liq correspond respectively to the density of supersaturated vapor and liquid densities, γ is the superficial tension and m is the molecular mass. The density values were calculated with the Monte Carlo simulations using the Gibbs ensemble, including the execution of 104 cycles, and each cycle with 25 movements distributed as follows: ten (10) translations, five (5) molecules re-growing, five (5) volume changes and five (5) transfers (insertions/eliminations) of molecules. Due to the difficulties to move insertions with large molecules such as hexane or nonane because of the low probability of acceptance, [30] a biased statistical model with flexible molecule, called Configurational Bias by Monte Carlo - CBMC, was included allowing to improve the acceptance probability [29].
The simulations with the Gibbs ensemble considered the definition of two simulation boxes: one cubic box side L, ranging from 100 and 300 A with N molecules mainly with light components (methane, ethane, etc., possible gas phase) [30]. The other box took into account box sides L between 20 and 40 Å, with N molecules mainly with dense components (pentane, hexane, nonane, etc.,). The density relation (kg/m3) and the length of the box L was: p=N*m/N A L3 where m corresponds to the molecular weight and N A is the Avogadro number.
The superficial tension γ was estimated using the SRK EOS [67]. The nucleation velocities were calculated for the simulations with "Heptane" and "Methane/Nonane" cases and compared with the results reported by Chen et a¡ [32] and Luijten [5], respectively. The nucleation velocity results for heptane is shown in Figure 3.
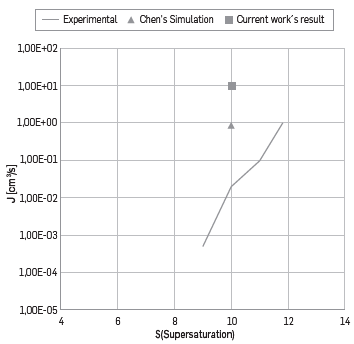
Figure 3 Heptane nucleation velocity. Continuous line: experimental values [68], triangle: reported results for Chen et al. [32]. Squared: results of the current work.
Figure 3 shows only one value derived from the current work, due to computational limited resources. However, results comparison suggests that the estimated nucleation velocity in the current work, is three magnitude orders greater than the one reported experimentally [68] and comparable to the obtained results by Chen et a¡ [32]. It is important to highlight that the main disagreement with the experimental tests, is based upon the latter been taken at 240 K, with concentrations lower than 0.001mol/mol of condensable components, due to difficulties in the measurement [18]; this leads to supersaturation values between 6 and 30.
On the other hand, results for the nucleation velocity of the system "Methane/Nonane" are shown in Figure 4, including experimental results obtained by Luijten [5]. In this case, computational results show a deviation of approximately two magnitude orders with respect to the experimental results; this deviation is in agreement with different reports that show deviations up to 5 orders of magnitude [18];[32]. The deviations might be due to the values for superficial tension and for the supersaturation. Additional studies are required to obtain appropriate values for these properties.
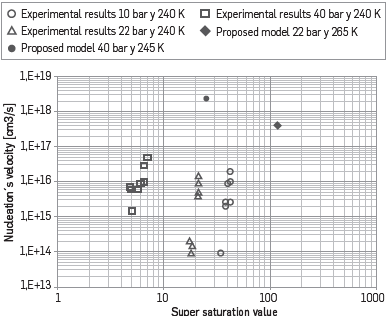
Figure 4 Comparison of obtained results with the proposed model and experimental results taken from [5].
CALCULATION OF THE DROPLET GROWTH.
An evaluation of the K n for the used systems was conducted for the selection of the appropriate model to use for modelling the droplet growth. K n values were found to be 0.002 for the "Methane/Nonane" system and 0.0006 for the "Natural Gas" system. According to the values for Kn, the adequate model for the calculation of growth corresponds to the Young model. The solution to the equations derived from Young's model was obtained through the Software Matlab®; the entry defined data for the system corresponds to the density for the number of drops and the size of the nucleus previously obtained by the relevant simulations of Monte Carlo simulations. The results obtained from the solution of Young's model for the mix Methane/Nonane at 40 bar and 240 K are reported in Table 2.
Table 2 Results of the cluster’s size and of the drop’s size obtained for each model and experimented for the mix metane/nonane at 40 bar.
| Model | Cluster's size [um] | Density of the number of the cluster [# of molecules/m3] | Drop's size [um] | Time [ms] ■ |
| Experimental result ([5)) | -- | -- | 0.21 | 23 |
| Results of the current work. | 0.0007 | 7.14E+05 | 0.56 | 23 |
| TNC and Young results | 0.009 | 6.20E+09 | 1,45 | 23 |
| Result using GPSA | -- | -- | 0.05 to 10 | -- |
| Result using the Kataoka model. | -- | -- | 12 | -- |
According to Table 2, the drop size as predicted by this work is higher than the size reported experimentally [5], with a difference of approximately 0.35 μm. Table 2 also shows the estimated sampled drop's sizes using the TNC and the Young method; the drop size with this method is 0.84 μm higher than the experimental value. As mentioned, the TNC is the right one for the analysis of pure substances but for multicomponent mixtures it may show considerable deviations. The deviations might be attributed to macroscopic approximations for the physical properties. As per the above, it is important to clarify that the application in this study of the TNC (for comparison purposes) was based upon the theory of the multicomponent nucleation described by Wilemski [65] and other considerations provided by Looijmans et al. [66]. For the calculation of the required macroscopic properties for the equation of the SRK EOS[67] and to relate the vapor pressure to the liquid, a Laplace relation was used, calculating the superficial tension using the model proposed by Hubbard [69].
Likewise, Table 2 shows the estimated values of the drop size using the application of the empiric models GPSA -[70] and [71] used in the design and the evaluation of industrial separators. From the comparison of predictions made by the different models, the model applied in the current work, based upon Young's theory together with the simulated nucleation of Monte Carlo, reports the closest value to the experimentally measured data.
For the "Natural Gas" system, Table 3 provides the derivate values of the application of the different models. According to this Table, the model applied in the current study (molecular simulation + Young's model) shows a difference of ca. 1.1 μm for the size of the drop, with respect to the experimental value. By contrast, the reported results by other models (TNC, GPSA and Kataoka), show higher deviations. As for the previous system, the model based upon the Monte Carlo simulation for the detection of critical nucleus corresponds to an adequate option providing a closeness to the experimental values; the differences with the experimental values could be due to the approximation for the composition of the systems used in molecular simulations.
Table 3 Results of the cluster and drop size obtained from each model and experiment for the mix of "Natural Gas" at 22 bar.
| Model | Cluster's size [μm] | Density of the cluster's number 1/m3 | Drop's size [μm] |
| Luijten experimental result | -- | -- | 0,85 |
| Current study result | 0.000695 | 2.02E+13 | 2.09 |
| TNC and Young results | 0.00151 | 4.20E+12 | 3.67 |
| Result using GPSA | -- | -- | 0.05 to 10 |
| Kataoka empirical result | -- | -- | 15 |
CONCLUSIONS
The current work was focussed upon the prediction of the drop size formed within the process of condensation of the natural gas, considering the two current stages in this phenomenon: nucleation and droplet growth. With this study, it was possible to carry out the gas-liquid nucleation through the molecular simulation using the Monte Carlo method with configurational bias; the simulation Includes the alkane chain configuration, the umbrella sampling technic for the calculation of the energy barrier and the tWF model for the detection of critical nucleus. It was possible to conduct simulations considering the different compositions of the natural gas (a mix of up to six components) at 40 bar with the isobaric-isothermal ensemble (NPT) considering
The results of the nucleation for the "Heptane" system reported a difference of three orders of magnitude with respect to the experimental data; however, this deviation is shown in the different works, displaying differences between 2 and 10 orders of magnitude. For the case of mix Methane/Nonane, one deviation of three orders of magnitude was found between the results of the velocity of nucleation obtained in this work, in respect of the ones reported by Luijten [5] at pressures between 10 and 40 bar. Deviations might be due to the used method for the detection of nucleus and windows used in the umbrella testing.
The application of the Young's growth drop model was used for the mix of methane with nonane between 10 and 40 bar, resulting in a maximum drop radios of 0,5 μm with a positive difference of 0,3 μm with respect to the experimental results. This is an important result as it gets closer better to the experimental values in comparison to the predicted values through empiric methods (used in the industry), based on experimental real conditions of separation. At the same time a better prediction for the size of the condensate drop, will allow to design separators gas-liquid more efficiently. The result of the current work are susceptible to be improved, for example through the estimation of the properties of fluids as the superficial tension using a molecular simulation.

