










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkUniversitas Scientiarum
Print version ISSN 0122-7483
Univ. Sci. vol.13 no.3 Bogotá Sept./Dec. 2008
Antimaláricos: construcción de híbridos moleculares de la cloroquina
Antimalarials: construction of molecular hybrids based on chloroquine
Antimaláricos: Construção de híbridos moleculares da cloroquina
kouznet@uis.edu.co
Recibido: 19-05-2008; Aceptado: 29-01-2009
Resumen
La resistencia desarrollada por el Plasmodium falciparum ante los fármacos tradicionales del tipo 4-amino-7-cloroquinolínicos ha llevado al diseño y síntesis de inhibidores duales, como los híbridos de la cloroquina. Además del farmacóforo típico en esta clase de antimaláricos, tales híbridos incorporan un espaciador metilénico como parte de un fragmento diamínico, cuyo propósito es la funcionalización del grupo amino terminal en sistemas heterocíclicos diversos con actividades biológicas reportadas. Esto con el fin de evitar la rápida e liminación de la molécula por parte del parásito, lo que se conoce como resistencia. Se discuten el diseño y síntesis de estos híbridos moleculares.
Palabras clave: malaria, resistencia a la cloroquina, híbridos moleculares, funcionalización.
Abstract
The resistance developed by Plasmodium falciparum against traditional 4-amino-7-chloroquinolinic drugs has lead to the design and synthesis of dual inhibitors such as chloroquine hybrids. Besides the usual pharmacophore in this type of antimalarials, such hybrids incorporate a methylenic spacer as part of a diaminic fragment whose purpose is the functionalization of the terminal amino group in various heterocyclic systems having reported biological activities. The prime goal is to avoid the molecule rapid elimination by the parasite, which is known as resistance. The design and synthesis of these molecular hybrids are discussed.
Key words: malaria, chloroquine resistance, molecular hybrids, functionalization.
Resumo
A resistência desenvolvida pelo Plasmodium falciparum frente aos fármacos tradicionais do tipo 4-amino-7-cloroquinolínicos tem levado ao desenho e síntese de inibidores duais, como os híbridos da cloroquina. Além do farmacóforo típico nesta classe de antimaláricos, tais híbridos incorporam um espaçador metilénico como parte de um fragmento diamínico, que tem por propósito a funcionalização do grupo amino terminal em sistemas heterocíclicos diversos com atividades biológicas reportadas. Isto com a finalidade de evitar a rápida eliminação da molécula por parte do parasito, o que se conhece como resistência. É discutido o desenho e sínteses destes híbridos moleculares.
Palavras-Chave: malária, resistência à cloroquina, híbridos moleculares, funcionalização.
Introducción
Diseño y síntesis de nuevos agentes antiparasitarios utilizando un producto natural como prototipo son tareas importantes de la química medicinal. Uno de los ejemplos más sobresalientes involucra a los indígenas suramericanos, quienes extraían la corteza de los árboles Cinchona, usando el extracto para combatir los escalofríos y la fiebre en el siglo XVII. En 1633 se introdujo esta medicina herbal en Europa, donde se le dio el mismo uso y también contra la malaria. En 1820 se aisló el componente activo, determinándose posteriormente que se trataba de la quinina (1), el primer compuesto en exhibir actividad antimalárica significativa. Para eliminar sus efectos tóxicos, se intentó mantener, sintéticamente, algunas de las características químicas y estructurales de este alcaloide usándolo como compuesto- líder, así se produjo el análogo sintético, la cloroquina (CQ) (2), que fue, inicialmente, un tratamiento excelente para la malaria (Figura 1) (Woster, 2001).
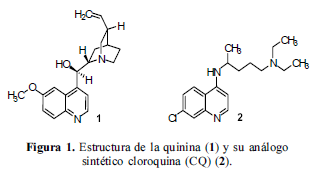
El paludismo es una enfermedad que puede causar la muerte en pocas horas, tan prevalente que en algunas zonas prácticamente todos los niños la han padecido durante el primer año de vida. Según la Organización Mundial de la Salud, en su reporte sobre la malaria en el mundo 2005, el paludismo es la causa de muerte de más de un millón de personas anualmente y constituye un riesgo para 3.200 millones de personas que viven en 107 países y territorios. Los niños menores de 5 años representan, con mucho, la mayor parte de las defunciones. El 14% de la población de las Américas vive en situación de riesgo, pero la parte de defunciones de esta región en el total mundial es mínima. Colombia es uno de los países de alta incidencia del protozoario Plasmodium falciparum, con 160.000 casos reportados en 2003 (World malaria report 2005; Carmona-Fonseca, 2007). La quinina y su prototipo simple - la CQ, son fármacos importantes y actuales en la lucha contra la malaria causada por los protozoos del género Plasmodium (Wiesner et al., 2003). La resistencia de este parásito frente a los fármacos antimaláricos actuales (sobre todo, la cloroquina) obliga a identificar nuevos compuestos alternativos y a diseñar los inhibidores duales (fármacos dobles) o híbridos, que podrían inhibir la formación de la hemozoina y otra diferente diana de este protozoario. Aún más, nuevos derivados e híbridos basados en el esqueleto de la CQ (7-cloro-4-aminoquinolinas) podrían combatir protozoos de diversos géneros, causantes de las enfermedades parasitarias leishmaniasis y tripanosomiasis americana (enfermedad de Chagas), entre otras. Trabajando varios años con las moléculas quinolínicas, nuestro laboratorio (http://ciencias.uis.edu.co/labqobio) ha generado nuevas bibliotecas pequeñas de diferentes clases de heterociclos quinolínicos, los cuales poseen buena actividad antiparasitaria (Kouznetsov et al., 2004; Kouznetsov, Rodríguez et al., 2004; Kouznetsov et al., 2006; Kouznetsov et al., 2007). Continuando esta actividad, en nuestro laboratorio se inició el estudio sintético hacia nuevos y propios híbridos de la CQ, cuyos resultados interesantes nos obligan a explorar más este tema tan promisorio e importante en la búsqueda de nuevos agentes antimaláricos (Amado Torres, 2008; Vargas Méndez et al., 2007).
1.1. Ciclo de vida del parásito de la malaria Plasmodium falciparum
Con la picadura de un mosquito Anofeles hembra infectado, los esporozoitos se transfieren a la corriente sanguínea humana (A) (Figura 2). Los esporozoitos invaden las células hepáticas e inician su división asexual, que resulta en la producción de varios miles de merozoitos (B). Los merozoitos se liberan desde las células hepáticas para infectar los eritrocitos en la corriente sanguínea (C). Una vez dentro de los eritrocitos, la reproducción asexual ocurre en ciclos de 48 horas (D) (Vickerman, 2005). Los parásitos se desarrollan en etapas anulares, tropozoitos y luego en esquizontes. En la etapa de segmentación, cada esquizonte se divide, típicamente, en 16 merozoitos eritrocíticos, los cuales se liberan por lisis del eritrocito e inmediatamente invaden nuevos eritrocitos.
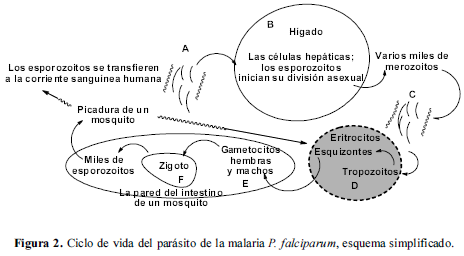
Una pequeña parte de los parásitos en la etapa sanguínea sufre diferenciación en gametocitos hembras y machos (E), los cuales entran en el mosquito cuando éste pica a una persona infectada. En el intestino del mosquito, los gametocitos hembras se desarrollan en macrogametos y los gametocitos machos se dividen de 4 a 8 microgametos flagelados. Los gametos hembra y macho se fusionan formando un zigoto (F). Éste último se transforma en un oocineto móvil que penetra en la pared del intestino y se convierte en un oocisto, residente bajo la membrana externa del intestino medio del mosquito. La división asexual dentro del oocisto produce miles de esporozoitos, liberados al romperse el oocisto, migrando luego hacia las glándulas salivares (Good, 2005).
1.2. Desarrollo de las aminoquinolinas
A pesar de su baja eficacia y tolerabilidad, la quinina aun juega un rol importante en el tratamiento de la malaria multirresistente debido a su alta solubilidad y porque se puede suministrar intravenosa en pacientes que ya no pueden tolerar la medicación oral. La pamaquina (3) (Figura 3), sintetizada en 1925, es una 8-aminoquinolina y uno de los primeros antimaláricos sintéticos que resultó ser más potente que la quinina al erradicar las etapas hepáticas del parásito en humanos y el resurgimiento de la malaria por Plasmodium vivax. En 1932 se desarrolló la mepacrina (4) (Figura 3), la cual es activa contra las etapas sanguíneas del P. falciparum, también es conocida como atebrina o quinacrina y recordada de la Segunda Guerra Mundial por la pigmentación amarilla que producía en la piel de los soldados (Foley y Tilley, 1998).
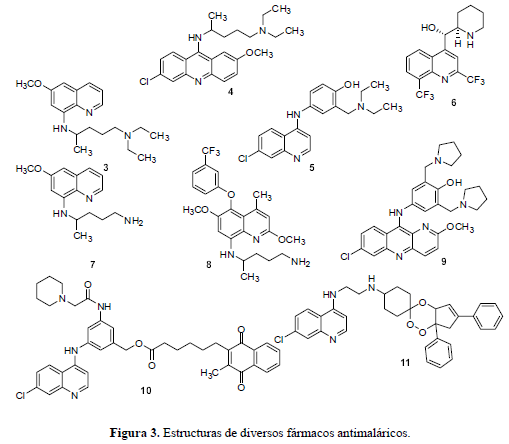
1.3. Otros compuestos antimaláricos
Entre los compuestos activos más eficaces de la actualidad se encuentra el sesquiterpeno artemisinina (12) (Figura 4), un 1,2,4,-trioxano, el potente componente antimalárico de la ancestral Artemisia annua, empleada como remedio herbal en China para la fiebre y más recientemente para el tratamiento de P. falciparum multirresistentes. Sin embargo, su valor terapéutico disminuye debido a su pobre solubilidad; como consecuencia se han desarrollado varios derivados. Por reducción de la artemisinina se obtiene la dihidroartemisinina (13), a partir de la cual se llega a una serie de derivados semisintéticos de primera generación: artemeter (14) y arteter (15) (Figura 4), compuestos más potentes que la artemisinina pero más tóxicos para el SNC y con menor tiempo de vida en el plasma, rápidamente excretados en la orina (Biagini et al., 2003).
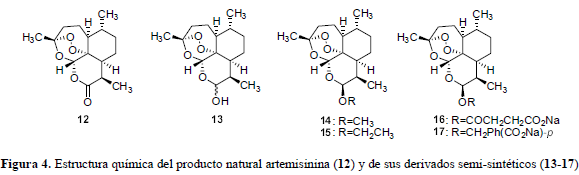
Como una nueva alternativa se desarrollan el artesunato de sodio (16) y el artelinato de sodio (17), con menor toxicidad en el SNC. Este tipo de moléculas representan una nueva clase de compuestos antimaláricos que no se basan en la estructura de la quinolina y que además son activos contra las cepas multirresistentes, sin surgimiento de resistencia alguna hasta el momento, convirtiéndose rápidamente en el fármaco de elección en la mayoría de los casos de malaria en los países donde se presenta y constituyéndose en la clase de antimalárico más importante disponible actualmente (Krishna et al., 2004). Se ha podido establecer que la unidad de peróxido de estos trioxanos es esencial para su potencia como antimalárico. El entendimiento del mecanismo de acción y el metabolismo de la artemisinina (12) y los endoperóxidos semisintéticos (13-17) (Figura 4) es un objetivo esencial para el desarrollo de nuevos trioxanos antimaláricos (Posner y O'Neill, 2004).
1.4. Análogos de la cloroquina
Desde los años cuarenta los análogos de la CQ (2) se han preparado de la misma forma y buscando el mismo objetivo: mejorar la actividad antimalárica (Carmack et al., 1946). Para tal propósito se han elaborado innumerables librerías de compuestos que han servido para enriquecer el conocimiento sobre los factores que afectan la actividad biológica de este tipo de moléculas (Singh et al., 1969. Singh et al., 1971). También durante el transcurso de este nuevo siglo, los químicos orgánicos sintéticos siguen trabajando con los análogos de la CQ (18-21), incorporando características novedosas en el grupo amino terminal, en la cadena carbonada espaciadora o incluso, buscando alternativas singulares que incluyan el núcleo 7-cloroquinolínico. Entre estas variaciones se encuentran análogos que sustituyen la función dietilamina por cadenas más cortas con grupos amino (Figura 5).
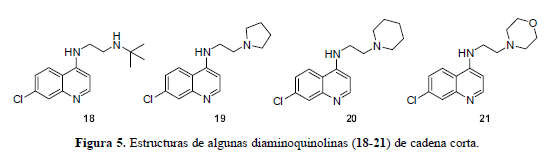
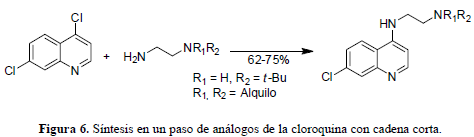
Las cadenas cortas con grupos alquilamino terminales secundarios y terciarios en lugar de la cadena de isopentileno en la cloroquina, le dan mayor elasticidad para propósitos metabólicos. De hecho, en los ensayos in vitro, estos análogos resultaron ser sustancialmente más potentes que el mismo fármaco e incluso inhibieron el crecimiento de las cepas resistentes en el rango nanomolar, lo cual es consecuencia de remplazar el grupo dietilamino con un grupo básico metabólicamente inerte como lo puede ser el t-butilo, el N-piperidinilo o el N-pirrolidinilo, mostrando mejores resultados el compuesto con t-butilo (18) y una reducción significativa en la actividad, el que tiene el grupo N-morfolinilo (21). Los anteriores análogos (18-21) se sintetizan en un paso único a partir de materiales dispuestos para esta reacción (Figura 6) (Ward et al., 2002).
La variación en la longitud de la cadena carbonada (espaciador molecular) y la gran diversidad que se puede incorporar con los sustituyentes del grupo amino terminal permiten generar librerías de compuestos quinolínicos, útiles en el descubrimiento de moléculas activas contra la malaria.
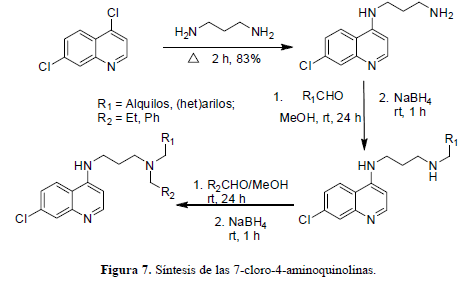
Los métodos sintéticos que permiten el acceso a grupos aminos sustituidos son de gran interés para la química medicinal, ya que las cadenas laterales alquilamínicas se encuentran en diversos fármacos. Se ha desarrollado un método sintético que permite la introducción de dos puntos de diversidad diferentes en el grupo amino lateral por aminaciones reductivas indirectas y secuenciales. Con los aldehídos comercialmente disponibles y la ruta sintética de la figura 7 se pueden llegar a elaborar librerías de 300.000 productos teóricos posibles que al ser "filtrados", para remover candidatos que posean átomos del tipo no orgánico o subestructuras reactivas o compuestos que no obedezcan la "Regla de Cinco" de Lipinski, es decir, moléculas demasiado grandes o pesadas para los estándares de los fármacos, se obtienen alrededor de 850 productos cuyo análisis retro-sintético lleva a 97 aldehídos precursores (Guy et al., 2004).
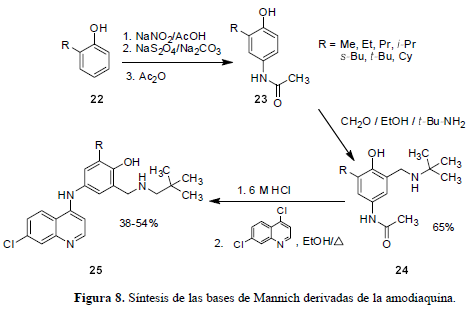
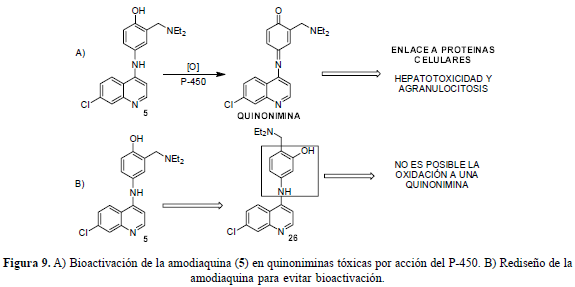
En la búsqueda de compuestos que evadan el mecanismo de resistencia que emplea el parásito, cualquiera que éste sea, pero que mantengan la actividad antiplasmodium, se han transformado los análogos ya existentes de la CQ (2) como la amodiaquina (5), una base de Mannich, con el propósito de establecer las características estructurales que deben tener estos antimaláricos. Los derivados de la base de Mannich remplazan la función dietilamina de la amodiaquina (5) por un grupo N-t-butilamino para prevenir que los compuestos con cadenas laterales se metabolicen aumentando la resistencia cruzada. La ruta sintética para llegar a estas bases de Mannich alquil sustituidas se diagrama en la figura 8 se inicia con la alquilación de un fenol (22) vía la formación de los compuestos (23) y (24), cuya hidrólisis y tratamiento con la 4,7-dicloroquinolina comercial, en etanol, produce moléculas (25) con rendimientos del 50%, con buenos resultados de inhibición in vitro contra cepas resistentes a la CQ (Ward et al., 1999).
Sin embargo, estos derivados, al poseer un hidroxilo en posición para con respecto al grupo amina, sufren oxidación por el citocromo P-450 y se transforman en quinoniminas, moléculas químicamente más reactivas. La amodiaquina y sus derivados 4'-hidroxilados se oxidan a esta clase de metabolitos (figura 9A), cuya formación in vivo y su enlace subsiguiente a las macromoléculas celulares podrían afectar la función celular directamente o por mecanismos inmunológicos, produciendo hepatotoxicidad o agranulocitosis. Se ha demostrado que la presencia del grupo hidroxilo en la posición 4' imparte gran actividad antimalárica contra los parásitos resistentes, en comparación con sus análogos deshidroxilados. El intercambio entre el grupo hidroxilo y la cadena lateral de Mannich provee una manera de prevenir la oxidación a metabolitos tóxicos, mientras se retienen las interacciones de los posibles enlaces importantes con el hidroxilo aromático (figura 9B) (O'Neill et al., 2003). El prototipo isoquina (26), es un regioisómero de la amodiaquina (5) que no puede formar metabolitos tóxicos por simple oxidación y que sigue siendo activo contra los parásitos resistentes in vitro.
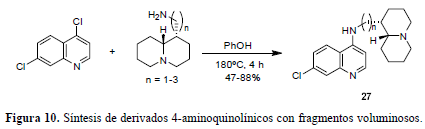
Una de las estrategias más prometedoras y exitosas en la lucha contra la malaria es la quimioterapia combinada, en ella se usa un derivado de la artemisinina (12) (figura 4) junto con un antimalárico convencional (4-aminoquinolina). Su propósito es mejorar la eficiencia y retrasar el surgimiento de resistencia. A la luz de las observaciones anteriores, junto con la resistencia que generan los compuestos análogos con cadenas laterales dialquilaminoalquílicas y los compuestos análogos metabólicamente más estables, con actividad antimalárica incrementada (producto de poseer cadenas laterales con átomos de nitrógeno básicos en un anillo piperidínico o pirrolidínico) se sintetizan y estudian derivados 4-aminoquinolínicos caracterizados por la presencia de fragmentos bicíclicos voluminosos, fuertemente básicos y lipofílicos (27) (figura 10), como el anillo de la quinolizidina, que se supone difícil de metabolizar (Sparatore et al., 2005). Estos compuestos se preparan por reacción entre la 4,7- dicloroquinolina con 1-aminoquinolicidina en presencia de fenol (figura 10) y muestran actividad entre 5 y 10 veces superior a la de la CQ en algunas cepas. El fragmento voluminoso y básico característico resulta ser interesante.
Se ha notado que varios análogos que contienen un enlace de hidrógeno intramolecular en alguno de sus fragmentos constituyentes, fueron activos contra P. falciparum multirresistentes, lo cual ha llevado a la exploración de cuan importante es esta característica. Para tal propósito, se sintetizó una serie de 116 compuestos con cuatro diferentes grupos alquilo y varios sustituyentes aromáticos, con la capacidad de formar enlaces de hidrógeno (figura 11).

1.5. Mecanismo de acción de las 4-aminoquinolinas
La CQ es activa sólo contra las etapas eritrocíticas del Plasmodium y, ciertamente, sólo contra aquellas etapas en las cuales el parásito está degradando activamente la hemoglobina. Además, también se ha asumido que la cloroquina podría interferir, de alguna forma, con el proceso de alimentación del parásito, llevándolo a su muerte por inanición. Las 4-aminoquinolinas se acumulan en altas concentraciones dentro de los compartimentos acídicos del parásito, lo cual es esencial para la habilidad de inhibir su crecimiento. La CQ es una base débil diprótica (pKa1=8,1; pKa2=10,2), en su forma no protonada puede atravesar las membranas de los eritrocitos invadidos y moverse con el gradiente de pH para acumularse en la vacuola (pH∼5,5) del invasor (figura 12), donde se cree que las 4-aminoquinolinas ejercen su acción antimalárica. Sin embargo, el mecanismo preciso no se ha podido establecer pero sí varias hipótesis ampliamente aceptadas (Foley y Tilley, 1997).
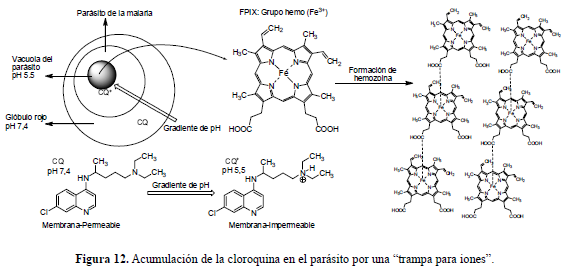
La CQ se acumula hacia abajo del gradiente de pH, de tal forma que su acumulación en el parásito es 10.000 veces mayor que en el glóbulo rojo. A medida que el parásito madura dentro del eritrocito invadido, digiere una gran cantidad de la hemoglobina (entre el 25% y 85% de la célula). La hemoglobina (Hb) ingerida es transportada hacia la vacuola del parásito. Una vez dentro de ésta, se inicia la digestión para proveer de nutrientes esenciales al invasor. La hemoglobinasa aspártica I inicia la degradación de la hemoglobina mientras que la hemoglobinasa aspártica II se enlaza a la hemoglobina desnaturalizada, debido al medio ácido, dando ferriprotoporfirina IX (FPIX) y globina. La tercera enzima involucrada es una cisteinproteasa (falcipaina) que no reconoce a la Hb ni a la FPIX, pero rápidamente se enlaza a la globina desnaturalizada, liberando un número de pequeños péptidos y aminoácidos que son esenciales para el crecimiento del parásito. El Plasmodium es incapaz de continuar degradando la FPIX libre y cualquiera sea la razón de su incapacidad, es claro que la FPIX no degradada es tóxica para el parásito, por lo que éste ha desarrollado un mecanismo para su desintoxicación. Para tal propósito, polimeriza la FPIX libre, formando una sustancia cristalina e insoluble conocida como hemozoina (β-hematina) o pigmento malárico, un complejo de coordinación no covalente con el hierro de una FPIX coordinando con el carboxilo de otra FPIX (O'Neill et al., 1998). Ya que tanto la liberación de los aminoácidos como la desintoxicación por el grupo FPIX por polimerización son esenciales para la supervivencia del parásito, ambos procesos podrían ser blancos de los medicamentos 4-aminoquinolínicos. La FPIX también es capaz de coordinar con bases nitrogenadas como las piridinas y las quinolinas. Entonces, las 4-aminoquinolinas podrían formar complejos con la FPIX libre impidiendo que el parásito la polimerizara para desintoxicarse, de hecho, la afinidad de la cloroquina por el parásito es similar a la afinidad de la FPIX por la cloroquina. Esta hipótesis se fortaleció al comprobarse que el complejo cloroquina-FPIX es incluso más tóxico para el Plasmodium que la FPIX libre. Esto, sumado a la elevada concentración que alcanza la cloroquina dentro de la vacuola, miles de veces superior, comparada con su concentración en las células eritrocíticas, hacen de las 4-aminoquinolinas compuestos interesantes para explorar las razones que han llevado a la resistencia hacia el medicamento cloroquina. Varios estudios han mostrado que la estructura del complejo formado entre la quinolina y la FPIX está dada por interacciones π y que tal estructura consiste de una molécula de quinolina y dos de FPIX, las cuales se aproximan a manera de sándwich. Los fragmentos tóxicos de FPIX, subproducto en la degradación de la Hb, se polimerizan en hemozoina insoluble. La CQ se acumula dentro de la vacuola y se cree que inhibe directamente la polimerización del grupo hemo atrapándolo, lo cual lleva a la intoxicación del parásito dada su incapacidad para deshacerse de la FPIX por otro método (O'Neill et al., 1998; Foley y Tilley, 1998). Si al grupo FPIX se le permitiese acumularse dentro de la vacuola, su concentración llegaría a 300-500 mM. Este grupo es muy tóxico porque puede generar especies oxigenadas reactivas e inducir estrés oxidativo, llevando al parásito a la muerte (figura 13).
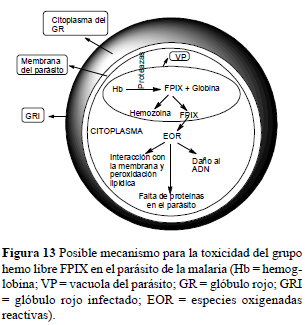
Como el grupo hemo (Fe3+) es una molécula lipofílica, se puede intercalar fácilmente en la membrana y causar cambios en su permeabilidad, organización lipídica e inducir peroxidación lipídica de la membrana. Lo anterior promueve la lisis de la célula y, eventualmente, la muerte del parásito. El grupo hemo libre también puede interferir con la degradación de la Hb; la cistein-proteasa, falcipaina, es muy sensible al grupo hemo, lo que podría culminar en muerte por inanición (Kumar et al., 2007). La gran diferencia de concentración de las 4-aminoquinolinas entre el parásito y el eritrocito hacen que la toxicidad del medicamento sea menor en comparación con otros compuestos, algunos de ellos también quinolínicos; sin embargo, la relevancia fisiológica de estos compuestos continúa observándose muy de cerca (Kwiek et al., 2004).
1.6. El rol del grupo en la posición C-7 del anillo quinolínico
Buscando los componentes estructurales necesarios para conseguir el nuevo agente antimalárico que supere los problemas de resistencia y mantenga una excelente bioactividad y baja o nula toxicidad, se ha encontrado que 7-bromo y 7-yodo aminoquinolinas con cadenas laterales diaminoalcánicas cortas (2 ó 3 carbonos) y largas (10-12 carbonos) son activas contra parásitos P. falciparum resistentes y sensibles a la cloroquina. Los resultados sugieren que el número de carbonos entre los dos nitrógenos de la cadena diaminoalcánica de las 7-bromo o las 7-yodo aminoquinolinas es crucial para la actividad contra los P. falciparum resistentes, al igual que con las aminoquinolinas 7-cloro sustituidas (Krogstad et al., 1998).
Las evidencias permiten proponer un detallado modelo de relación estructura-actividad para la CQ así: el núcleo de la 4-aminoquinolina provee, por sí solo, una estructura acomplejante del grupo hemo, pero no lo suficiente para la inhibición de la formación de hemozoina; el cloro en la posición 7 es responsable por la inhibición en la formación de hemozoina pero, probablemente, tiene poca influencia en la fuerza de asociación con el hemo; la cadena lateral aminoalquílica es un requerimiento para la fuerte actividad antiplasmódica, probablemente ayuda con la acumulación en la vacuola y también parece aumentar la fuerza de asociación con el hemo en algunos casos (figura 14) (Egan et al., 2000).
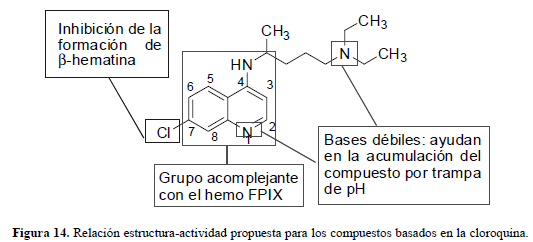
Los cambios en la longitud de la cadena lateral diaminoalquílica tienen poca influencia sobre la actividad contra las cepas sensibles a la CQ, pero una influencia profunda contra las cepas resistentes. Parece que con tan solo hacer grandes cambios en tal cadena lateral se puede superar la resistencia a la CQ, sin tener que hacer cambios en el fragmento 4-amino-7-haloquinolínico, responsable del acomplejamiento con el grupo hemo y la inhibición de la formación de β-hematina, lo cual se correlaciona con la capacidad electroatrayente del grupo en la posición C-7 del sistema quinolínico. Por su tamaño y propiedades, el grupo 7-cloro se ha identificado como una característica necesaria para la inhibición en la formación de β-hematina sobre otros grupos electroatrayentes o electrodonantes. Sin embargo, se propone como una característica necesaria pero no suficiente para una fuerte actividad antiplasmodium. Esto puede indicar que una reducción en la densidad electrónica de las posiciones del anillo quinolínico 5 u 8a, o ambas, es clave para la actividad. Aunque la razón no es clara es posible que ello permita que la quinolina asuma o mantenga una alineación o conformación, si cabe, particular con respecto a la hematina (Egan, et al., 2002). El pKa1 del nitrógeno quinolínico es fuertemente dependiente de la naturaleza del sustituyente en la posición 7 del anillo. Grupos electrodonantes como NH2 y OCH3 aumentan el pKa1, con relación al sustituyente H. Por el contrario, grupos fuertemente electroatrayentes como el NO2 causan un decrecimiento considerable en la basicidad por razones de resonancia en el anillo. Lo inesperado es la fuerte variación en el pKa2 del grupo amino terciario de la cadena lateral. Evidentemente, en los análogos de la CQ con cadenas laterales cortas, hay una interacción significativa a través del espacio entre el amino terminal de la cadena lateral y el anillo quinolínico, la cual disminuye, probablemente, al aumentar la longitud de la cadena. De lo anterior depende que el compuesto se acumule lo suficiente en la vacuola ácida y por lo tanto, se mantenga o disminuya la actividad (Egan, et al., 2002). En la identificación de compuestos antimaláricos 4-aminoquinolínicos más fuertes, solubles y con biodisponibilidad oral, se han desarrollado modelos farmacofóricos que ayudan en la búsqueda multidisciplinaria de moléculas más eficaces que la CQ, en teoría (Dascombe et al., 2005).
1.7. Híbridos de la cloroquina
La estrategia de hibridación también se utiliza para tratar de contrarrestar la resistencia en los parásitos. Con ella se crean compuestos quiméricos o híbridos que reúnen lo mejor de las propiedades de al menos dos moléculas diferentes, en el caso de los híbridos de la cloroquina, se trata del fragmento de 7-cloro-4-aminoquinolina, reconocido como crucial para las interacciones π-π con el grupo hemo FPIX dentro de la vacuola del Plasmodium, es decir, es el farmacóforo de este tipo de moléculas. El otro fragmento, generalmente heterocíclico, debe poseer también alguna clase de actividad biológica. En el caso de los híbridos de la CQ, la actividad está relacionada con contrarrestar la rápida difusión que sufren las 4-aminoquinolinas desde la vacuola y por ende, la disminución en su acumulación dentro del parásito. Hoy en día, los trabajos sintéticos con este enfoque se intensifican. La síntesis de los compuestos (29) es un ejemplo de ellos (figura 15) (Chibale et al., 2004).

Estas aminoquinolinas a-acilamino amidas son activas contra cepas de P. falciparum sensibles a la CQ y contra algunas resistentes a la CQ. Esta aproximación es análoga a la terapia combinada convencional en la cual se suministran dos o más medicamentos antimaláricos (cócteles). La misma táctica sintética se puede diseñar de tal forma que el producto final esté enfocado hacia múltiples blancos dentro del parásito. Introduciendo así variables cíclicas como las γ- y δ-lactamas e intentando disminuir los niveles de flexibilidad asociados con las amidas (29) así como también su biodisponibilidad (Veber et al., 2002). La reacción multi-componente de diaminas, con los ácidos levulínico (m = 1) ó 4-acetilbutírico (m = 2) (30) y los isonitrilos (31) t-butilnitrilo o ciclohexilonitrilo en metanol genera las lactamas (32) (figura 16) (Chibale et al., 2006).
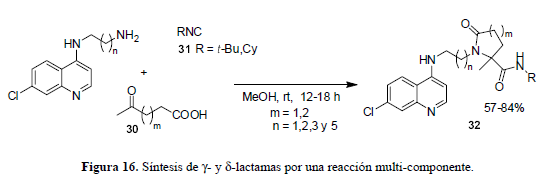
En general, los compuestos δ-lactámicos (m = 2) resultaron más activos que los γ-lactámicos (m = 1) aunque los resultados de actividad son modestos. Lo anterior puede ser debido a inhabilidad de estos compuestos para interaccionar con el grupo hemo o a que no logran llegar hasta el sitio de acción en la vacuola.
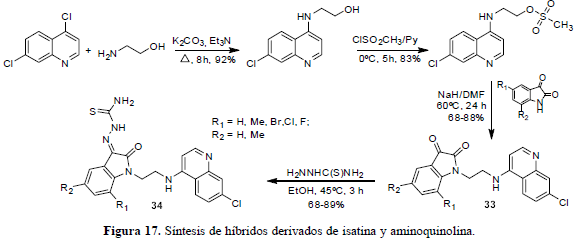
A los híbridos también se les denomina inhibidores duales o "fármacos dobles", por su potencial para actuar ante múltiples blancos dentro del mismo organismo o célula causante de la enfermedad. Se encuentra en la literatura el diseño y síntesis de híbridos derivados de isatina (Chauhan et al., 2005), un producto natural privilegiado sobre el que se pueden construir moléculas de mayor potencial, incluyendo 4-aminoquinolinas. En estos híbridos, la cetona y la tiosemicarbazona introducidas para proveer sitios reactivos (imina, carbotiol), sirven como ojivas electrófilas y además, ya tienen reportes sobre su actividad antiplasmodium (Chibale et al., 2005). Los híbridos (33) y (34) fueron preparados como en la figura 17. Las tiosemicarbazonas (34) mostraron mejores actividades contra tres cepas de P. falciparum y el potencial para inhibir la falcipaina-2.
Para tal efecto, las 4-aminoquinolin-semicarbazonas han resultado ser excelentes inhibidores de falcipaina-2 al ser combinadas estructuralmente con bases de Mannich. En la figura 18 estos híbridos (36) se preparan a partir del N1-(7- cloroquinolin-4-il)-1,2-diaminoetano (Chibale et al., 2007).

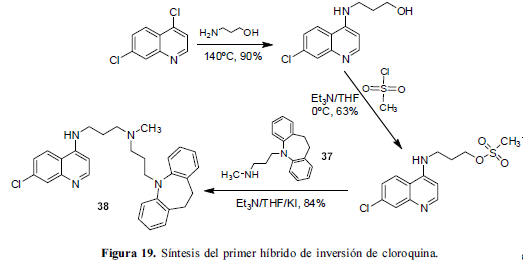
Se ha descubierto recientemente que la resistencia en los parásitos P. falciparum a la CQ está fuertemente asociada con mutaciones en una proteína de la membrana en la vacuola y como resultado de esta mutación se produce el transporte excesivo de CQ desde el sitio de acción en la vacuola hacia afuera de ésta. La proteína es la PfCRT (P. falciparum chloroquine resistance transporter), o transportador de resistencia a la CQ del P. falciparum. Se conoce de varias estructuras moleculares, denominadas agentes de inversión (reversal agents) que inhiben la PfCRT. Se propuso que enlazar un fragmento como el de la CQ a uno de estos agentes de inversión, para bloquear la eliminación de CQ de la vacuola, podría resultar en un híbrido altamente efectivo contra la malaria (Figura 19) (Peyton et al., 2006). Entre los agentes de inversión sobresalen el antidepresivo imipramina, uno de los más estudiados contra la PfCRT y su derivado, la des-N-metilimipramina (37).
Este fragmento del híbrido (38) no ocasiona detrimento en la actividad del farmacóforo cloroaminoquinolínico. De hecho, los resultados de IC50 contra las cepas resistentes Dd2 y sensibles D6 son mejores que los de la CQ, un hecho remarcable que encamina al híbrido (38) como compuesto- líder. La modificación selectiva del grupo amino terminal de las diaminoquinolinas con pequeños sistemas heterocíclicos podría modular la actividad antimalárica como parte de una estrategia para el desarrollo de antimaláricos. Las 4-tiazolidinonas son andamios privilegiados biológicamente y bien tolerados por el organismo humano, por lo que es apropiado estudiar la actividad antiplasmódica de sus híbridos, cuyos datos de actividad in vitro muestran actividad antimalárica potente y en algunos casos, los valores de IC50 son comparables o mejores que el de la CQ, lo cual prueba que este tipo de modificaciones en dicho átomo de nitrógeno son muy bien toleradas para propósitos de actividad antimalárica (Katti et al., 2007).
En conclusión, debido al costo y a la dificultad para obtener antimaláricos modernos, como son los derivados de artemisinina, los compuestos híbridos han resurgido en las últimas dos décadas como los más estudiados en la búsqueda de anti-Plasmodium que superen la resistencia generada por el parásito. Para alcanzar este objetivo es indispensable incorporar en tales estructuras al farmacóforo de la CQ y a grupos funcionales o sistemas heterocíclicos que eviten su rápida eliminación por el mecanismo de la proteína PfCRT.
Agradecimientos
Los autores presentan sus agradecimientos al Instituto Colombiano para el Desarrollo de la Ciencia y la Tecnología "Francisco José de Caldas" (COLCIENCIAS, proyecto CENIVAM, contracto 432-2004) por su constante apoyo financiero.
Literatura citada
AMADO TORRES, D.F. Construcción de híbridos moleculares de la cloroquina incorporando fragmentos alcanodiamínicos, vainillínicos y eugenilos. Tesis de maestría, UIS, 2008. [ Links ]
BIAGINI, G.A.; O'NEILL, P.M.; NZILA, A.; WARD, S.A.; BRAY, P.G. Antimarial chemotherapy: Young guns or back to the future? Trends in parasitology, 2003, 19: 479-487. [ Links ]
CARMACK, M.; BULLIT, O.H.; HANDRICK, G. R.; KISSINGER, L.W.; VON, I. The synthesis of 4-(4'-amino-1'-methylbutylamino)- 7-chloroquinoline and some 4-(4'-monoalkylamino- 1'-methylbutylamino)-7-chloroquinolines. Journal of the American Chemical Society, 1946, 68: 1220-1225. [ Links ]
CARMONA-FONSECA, J. Nuevos tratamientos para paludismo en Colombia, 2006. Acta Médica Colombiana, 2007, 32: 157-163. [ Links ]
CHAUHAN, P.M.S.; AGARWAL, A.; SRIVASTABA, K.; PURI, S.K. Synthesis of substituted indole derivatives as a new class of antimalarial agents. Bioorganic and Medicinal Chemistry Letters, 2005, 15: 3133-3136. [ Links ]
CHIBALE, K.; CHIPELEME, A.; GUT, J.; ROSENTHAL, P. Synthesis and biological evaluation of phenolic Mannich bases of benzaldehyde and (thio)semicarbazone derivatives against the cysteine protease falcipain-2 and a chloroquine resistant strain of Plasmodium falciparum. Bioorganic and Medicinal Chemistry, 2007, 15: 273-282. [ Links ]
CHIBALE, K.; CHIYANZU, I.; CLARKSON, C.; SMITH, P.J.; LEHMAN, J.; GUT, J.; ROSENTHAL, P. Design, synthesis and antiplasmodial evaluation in vitro of new 4-aminoquinoline isatin derivatives. Bioorganic and Medicinal Chemistry, 2005, 13: 3249-3261. [ Links ]
CHIBALE, K.; MUSONDA, C.C.; GUT, J.; ROSENTHAL, P.J.; YARDLEY, V.; CARVALHO DE SOUZA, R.C. Application of multicomponent reactions to antimalarial drug discovery. Part 2: New antiplasmodial and antitrypanosomal 4-aminoquinoline ã– and ä–lactams via a "catch and release" protocol. Bioorganic and Medicinal Chemistry, 2006, 14: 5605-5615. [ Links ]
CHIBALE, K.; MUSONDA, C.C.; TAYLOR, D.; LEHMAN, J.; GUT, J.; ROSENTHAL, P. Application of multi-component reactions to antimalarial drug discovery. Part 1: Parallel synthesis and antiplasmodial activity of new 4-aminoquinoline Ugi adducts. Bioorganic and Medicinal Chemistry Letters, 2004, 14: 3901-3905. [ Links ]
DASCOMBE, M.; DREW, M.; MORRIS, H.; WILAIRAT, P.; AUPARAKKITANON, S.; MOULE, W.; ALIZADEH-SHEKALGOURABI, S.; EVANS, P.; LLOID, M.; DYAS, A.; CARR, P.; ISMAIL, F. Mapping antimalarial pharmacophores as a useful tool for the rapid discovery of drugs effective in vivo: Design, construction, characterization, and pharmacology of metaquine. Journal of Medicinal Chemistry, 2005, 48: 5423-5436. [ Links ]
EGAN, T.J.; HUNTER, R.; KASCHULA, C.H.; MARQUEZ, H.M.; MISPLON, A.; WALDEN, J. Structure-function relationship in aminoquinolines: Effect of amino and chloro groups on quinoline-hematin complex formation, inhibition of â-hematin formation, and antiplasmodial activity. Journal of Medicinal Chemistry, 2000, 43: 283-291. [ Links ]
EGAN, T.J.; KASCHULA, C.; HUNTER, R.; BASILICO, N.; PARAPINI, S.; TARAMELLI, D.; PASINI, E.; MONTI, D. Structure-activity relationship in 4-aminoquinoline antiplasmodials. The role of the group at the 7-position. Journal of Medicinal Chemistry, 2002, 45: 3531-3539. [ Links ]
FOLEY, M.; TILLEY, L. Quinoline antimalarials: Mechanisms of action and resistance and prospects for new agents. Pharmacological Therapy, 1998, 79: 55-87. [ Links ]
FOLEY, M.; TILLEY, L. Quinoline antimalarials: Mechanisms of action and resistance. International Journal for Parasitology, 1997, 27: 231-240. [ Links ]
GOOD, M.F. Genetically modified Plasmodium highlights the potential of whole parasite vaccine strategies. Trends in immunology, 2005, 26: 295-297. [ Links ]
GUY, R.K.; MADRID, P.B.; WILSON, N.T.; DERISI, J.L. Parallel synthesis and antimalarial screening of a 4-aminoquinoline library. Journal of Combinatorial Chemistry, 2004, 6: 437-442. [ Links ]
KATTI, S.B.; SOLOMON, V.R.; HAQ, W.; SRIVASTAVA, K.; PURI, S.K. Synthesis and antimalarial activity of side chain modified 4-aminoquinoline derivatives. Journal of Medicinal Chemistry, 2007, 50: 394-398. [ Links ]
KOUZNETSOV, V.V.; VARGAS MÉNDEZ, L.Y.; TIBADUIZA, B.; OCHOA, C.; MONTERO PEREIRA, D.; NOGAL RUIZ, J.J.; PORTILLO, C.F.; GÓMEZ BARRIO, A.; BAHSAS, A.; AMARO-LUIS, J. 4-NAryl( benzyl)amino-4-hetarylbut-1-enes as building blocks in heterocyclic synthesis. 4. Synthesis of 4,6-dimethyl-5-nitro(amino)-2-pyridylquinolines and their antiparasitic activities. Archiv der Pharmazie, 2004, 337: 127-132. [ Links ]
KOUZNETSOV, V.; RODRÍGUEZ, W.; STASHENKO, E.; OCHOA, C.; VEGA, C.; ROLÓN, M.; MONTERO PEREIRA, D.; ESCARIO, J.A.; GÓMEZ BARRIO, A. Transformation of schiff bases derived from a-naphthaldehyde. Synthesis, spectral data and biological activity of new 3-aryl-2-(a-Naphtyl)-4-thiazolidinones and N-aryl-N-[1-(a-Naphthyl)but-3-enyl]amines. Journal Heterocyclic Chemistry, 2004, 41: 995-998. [ Links ]
KOUZNETSOV, V.V.; RIVERO, J.; OCHOA, C.; STASHENKO, E.E.; RENÉ MARTÍNEZ, J.; OCHOA, C.;MONTORO PEREIRA, D.; NOGAL RUIZ, J.J.; FERNÁNDEZ PORTILLO, C.; MUELAS SERRANO, S.; GÓMEZ BARRIO, A.; BAHSAS, A.; AMARO-LUIS, J. Synthesis and antiparasitic properties of new 4-N-benzylamino-4- heteroarylbut-1-enes. 5. Archiv der Pharmazie, 2005, 338: 32-37. [ Links ]
KOUZNETSOV, V.V.; VARGAS MÉNDEZ, L.Y.; LEAL, S.M.; MORA CRUZ, U.; CORONADO, C. A.; MELÉNDEZ GÓMEZ, C.M.; ROMERO BOHÓRQUEZ, A.R.; ESCOBAR RIVERO, P. Target- Oriented Synthesis of Antiparasitic 2-Hetaryl Substituted Quinolines based on imino Diels-Alder reactions. Letters in Drug Design & Discovery, 2007, 4: 293-296. [ Links ]
KRISHNA, S.; UHLEMANN, A.C.; HAYNES, R.K. Artemisinins: Mechanisms of action and potential for resistance. Drug Resistance Updates, 2004, 7: 233-244. [ Links ]
KROGSTAD, D.J.; DE, D.; KROGSTAD, F.M.; BYERS, L.D. Structureactivity relationships for antiplasmodial activity among 7-substituted 4-aminoquinolines. Journal of Medicinal Chemistry, 1998, 41: 4918-4926. [ Links ]
KUMAR, S.; GUHA, M.; CHOUBEY, V.; MAITY, P.; BANDYOPADHYAY, U. Antimalarial drugs inhibiting hemozoin (â-hematin) formation: A mechanistic update. Life Sciences, 2007, 80: 813-828. [ Links ]
KWIEK, J.J.; HAYSTEAD, T.A.; RUDOLPH, J. Kinetic mechanism of quinone oxidoreductase 2 and its inhibition by the antimalarial quinolines. Biochemistry, 2004, 43: 4538-4547. [ Links ]
MADRID, P.B.; LIOU, A.P.; DERISI, J.L.; GUY, R.K. Incorporation of an intramolecular hydrogen-bonding motif in the side chain of 4-aminoquinolines enhances activity against drug-resistant P. falciparum. Journal of Medicinal Chemistry, 2006, 49: 4535-4543. [ Links ]
O'NEILL, P.M.; BRAY, P.G.; HPWLEY, S.R.; WARD, S.A.; PARK, B.K. 4-Aminoquinolines. Past, present and future: A chemical perspective. Pharmacological Therapy. 1998, 77: 29-58. [ Links ]
O'NEILL, P.; MUKHTAR, A.; STOCKS, P.; RANDLE, L.; HINDLEY, S.; WARD, S.; STORR, R.; BICKLEY, J.; O'NEIL, I.; MAGGS, J.; HUGHES, R.; WINSTANLEY, P.; BRAY, P.; PARK, B. Isoquine and related amodiaquine analogues: A new generation of improved 4-aminoquinoline antimalarials. Journal of Medicinal Chemistry, 2003, 46 (23): 4933-4945. [ Links ]
PEYTON, D.; BURGESS, S.; SELZER, A.; KELLY, J.; SMILKSTEIN, M.; RISCOE, M. A chloroquine-like molecule designed to reverse resistance in Plasmodium falciparum. Journal of Medicinal Chemistry, 2006, 49: 5623-5625. [ Links ]
POSNER, G.H.; O'NEILL, P.M. Knowledge of the proposed chemical mechanism of action and cytochrome P-450 metabolism of antimalarial trioxanes like artemisinin allows rational design of new antimalarial peroxides. Accounts of Chemical Research, 2004, 37: 397-404. [ Links ]
SERGHERAERT, C.; RYCKEBUSH, A.; DEPREZ-POULAIN, R.; MAES, L.; DEBREU-FONTAINE, M.A.; MOURAY, E.; GRELLIER, P. Synthesis and in vitro and in vivo antimalarial activity of N1-(7-chloro-4-quinolyl)-1,4-bis(3-aminopropyl) piperazine derivatives. Journal of Medicinal Chemistry, 2003, 46: 542-557. [ Links ]
SINGH, T.; STEIN, R.G.; BIEL, J.H. Antimalarials. Unsaturation in the chloroquine side chain and antimalarial activity. Journal of Medicinal Chemistry, 1969, 12: 368-371. [ Links ]
SINGH, T.; STEIN, R.G.; HOOPS, J.F.; BIEL, J.H.; HOYA, W.K.; CRUZ, D.R. Antimalarials. 7-Chloro-4-(substituted amino)quinolines. Journal of Medicinal Chemistry, 1971, 14: 283-286. [ Links ]
SPARATORE, A.; BASILICO, N.; PARAPINI, S.; ROMEO, S.; NOVELLI, F.; SPARATORE, F.; TARAMELLI, D. 4-Aminoquinoline quinolizidinyl- and quinolizidinilalkyl-derivatives with antimalarial activity. Bioorganic and Medicinal Chemistry, 2005, 13: 5338-5345. [ Links ]
VARGAS MÉNDEZ, L.Y.; AMADO TORRES, D.F.; KOUZNETSOV, V.V. Búsqueda de nuevos inhibidores de la acetilcolinesterasa", Abstr. in I Latin American Meeting on Medicinal Chemistry, 22-25 de abril de 2007, Montevideo -Uruguay, p. 32. [ Links ]
VEBER, D.; JOHNSON, S.; CHENG, H.; SMITH, B.; WARD, K.; KOPPLE, K. Molecular properties that influence the oral bioavaiability of drug candidates. Journal of Medicinal Chemistry, 2002, 45: 2615-2623. [ Links ]
VICKERMAN, K. The lure of life cycles: Cyril Garnham and the malaria parasites of primates. Protist, 2005, 156: 433-449. [ Links ]
WARD, S.A.; O'NEILL, P.M.; STOCKS, P.A.; RAYNES, K.J.; BRAY, P.G.; PARK, B.K. Novel short chain chloroquine analogues retain activity against chloroquine resistant K1 Plasmodium falciparum. Journal of Medicinal Chemistry, 2002, 45: 4975-4983. [ Links ]
WARD, S.A.; RAYNES, K.J.; STOCKS, P.A.; O'NEILL, P.M.; PARK, B.K. New 4-aminoquinoline Mannich base antimalarials. 1. Effect of an alkyl substituent in the 5'-position of the 4-hydroxyanilino side chain. Journal of Medicinal Chemistry, 1999, 42, 2747-2751. [ Links ]
WIESNER, J.; HASSAN, J.; SCHLITZER, M. New antimalarial drugs. Angewandte Chemie. International Edition, 2003, 42: 5274-5293. [ Links ]
WOSTER, P.M. Annual reports in medicinal chemistry: New therapies for parasitic infection. Academic press, cap. 10, San Diego, 2001. [ Links ]
WORLD MALARIA REPORT 2005. En: http://rbm.who.int/wmr2005/ Marzo de 2007. [ Links ]

