










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkUniversitas Scientiarum
Print version ISSN 0122-7483
Univ. Sci. vol.17 no.1 Bogotá Jan./Apr. 2012
Influence of Mg2+ ions on the interaction between 3,5-dicaffeoylquinic acid and HTLV-I integrase
Influencia de los iones Mg+2 sobre la interacción entre el ácido 3,5- dicafeoilquínico y la integrasa del HTLV-I
Influência dos íons Mg2+ sobre a interação entre o ácido 3,5-dicafeoilquínico e a integrase do HTLV-I
Ángela Peña1*, Juvenal Yosa2*, Yesid Cuesta-Astroz1, Orlando Acevedo3, Leonardo Lareo†, Felipe García-Vallejo1
1Laboratorio de biología molecular y patogénesis. Departamento de ciencias fisiológicas. Escuela de ciencias básicas. Departamento de Salud. Universidad del Valle. Sede San Fernando. Cali, Colombia.
2Universidad Distrital Francisco José de Caldas. Facultad de ciencias ambientales. Ingeniería ambiental. Av. Circunvalar Venado de Oro., Bogotá, Colombia.
3Laboratorio de bioquímica computacional y estructural y bioinformática. Departamento de nutrición y Bioquímica. Facultad de ciencias. Pontificia Universidad Javeriana. Bogotá, D.C. Colombia.
*viviana.pena@gmail.com; juvenal.yosa@unibas.ch
Received: 18-11-2011; Accepted: 23-03-2012
Abstract
Objective. Using molecular simulation, we studied the influence of Mg2+ ions on the binding mode of HTLV-I Integrase (IN) catalytic domain (modeled by homology) with the 3,5- Dicaffeoylquinic Acid (DCQA). HTLV-I Integrase homology model was built using template-like crystallographic data of the IN catalytic domain solved for Avian Sarcoma Virus (VSA, pdb: 1VSD). Materials and methods. In order to analyze the role of Mg2+ in the interaction or coupling between 3,5-DCQA and Integrase, three models were created: i) in the absence of Mg2+ ions, ii) with a Mg2+ ion coordinated at Asp15 and Asp72 and iii) model with two Mg2+ ions coordinated at Asp15-Asp72 and Asp72-Glu108. Coupling force and binding free energy between 3,5-DCQA and HTLV-I IN were assessed in the three models. Results. The lowest docking score and free energy binding were obtained for the second model. Mg2+ ion strongly affected the coupling of the inhibitor 3,5-DCQA with HTLV-I catalytic domain of Integrase, thus revealing a strong interaction in the ligand-protein complex regardless of the ligand-catalytic interaction sites for all three models. Conclusion. Altogether, these results strengthen the hypothesis that the presence of one Mg2+ ion could enhance the interaction in the complex by decreasing free energy, therefore increasing the affinity. Moreover, we propose 3,5-DCQA as an important pharmacophore in the rational design of new antiretroviral drugs.
Key words: 3,5 -Dicaffeoylquinic Acid, Human T-Lymphotropic Type I (HTLV-1), Integrase (IN), Homology Model, Molecular Docking, Binding Free Energy, Mg2+ Ions.
Resumen
Objetivo: Usando simulación molecular, estudiamos la influencia de los iones Mg2+ en la interacción del dominio catalítico de la integrasa HTLV-I (IN) (modelado por homología) con el ácido 3,5-Dicafeoilquínico (DCQA). Materiales y métodos. El modelo por homología de la HTLV-I IN fue construido usando como molde la estructura cristalina de la IN del virus del sarcoma aviar (VSA, pdb: 1VSD). Para analizar el rol de los iones Mg2+ en la interacción con el DCQA y la integrasa, tres modelos fueron creados: i) en ausencia de iones Mg2+, ii) con un ion Mg2+ coordinado con Asp15 y Asp72 y iii) con dos iones Mg2+ coordinados con Asp15-Asp72 y Asp72-Glu108. Las fuerzas de interacción y la energía libre de unión entre el DCQA y la HTLV-I IN fueron calculadas en los tres modelos. Resultados. El puntaje más bajo en el docking y la menor energía libre fueron obtenidos para el modelo con un solo ion de Mg2+. El Mg2+ afecta fuertemente el acoplamiento del inhibidor DCQA con el dominio catalítico de la HTLV-I IN, revelando una fuerte interacción entre el complejo ligando-proteína que es independiente del sitio catalítico de los tres modelos usados. Conclusión. Los resultados sugieren que la presencia de un ion de Mg2 + podría incrementar la interacción en el complejo debido a la disminución de la energía libre, intensificando así la afinidad. Por lo que, proponemos al DCQA como farmacóforo para el diseño de drogas antiretrovirales.
Palabras clave: ácido 3,5-Dicafeoilquínico (DCQA). Integrasa linfotrópica-T humana Tipo I (HTLV-1), Integrasa (IN), Modelo por homología, Docking molecular, Energía libre de unión, Iones Mg2+.
Resumo
Objetivo. Usando simulação molecular, estudamos a influência dos íons Mg2+ na interação do domínio catalítico da integrase HTLV-I (IN) (modelado por homologia) com o ácido 3,5-dicafeoilquínico (3,5-diCQA). Materiais e métodos. O modelo de homologia da HTLV-I IN foi construído utilizando como fôrma a estrutura cristalina da IN de vírus do sarcoma aviário (VSA, pdb: 1VSD). Para analisar o papel dos íons Mg2+ na interação com o 3,5-diCQA e a integrase, três modelos foram criados: i) na ausência de íons Mg2+, ii) com um íon Mg2+ coordenado com Asp15 e Asp72 e, iii) com dois íons Mg2+ coordenados com Asp15-Asp72 e Asp72-Glu108. As forças de interação e a energia livre de ligação entre o 3,5-diCQA e a HTLV-I IN foram calculadas nos três modelos. Resultados. A pontuação mais baixa no docking e a menor energia livre foram obtidas para o modelo com um único íon de Mg2+. O Mg2+ afeta fortemente o acoplamento do inibidor 3,5-diCQA com o domínio catalítico da HTLV-I IN, revelando uma forte interação entre o complexo ligando-proteína que é independente do sítio catalítico dos três modelos utilizados. Conclusão. Os resultados sugerem que a presença de um íon Mg2+ poderia aumentar a interação no complexo devido à diminuição da energia livre, aumentando assim a afinidade. Assim, propomos ao 3,5-diCQA como farmacóforo para a fabricação de medicamentos antirretrovirais.
Palavras-chave: ácido 3,5-dicafeoilquínico (3,5-diCQA). Integrase linfotropica-T humana Tipo I (HTLV-1), integrase (IN), Modelo por homologia, Molecular Docking, Energia livre de ligação, íons Mg2+.
Introduction
Human T-Lymphotropic Virus Type 1 (HTLV-I) infection is currently a serious problem of public health in several areas of the world including Japan, some regions of Africa, the Caribbean coast and some South American countries, affecting more than 20 million of inhabitants worldwide (1- 3). Therefore, HTLV-I infection is regarded as a global epidemic problem (4, 5). Although most of the infected individuals remain asymptomatic for long periods of time, 2-3 % develop adult T-cell leukemia (ATL) (6) and about 1-2 % of the infected individuals display an evolution towards a neurodegenerative chronic condition termed HTLV-I Associated Myelopathy Tropical Spastic Paraparesis (HAM/ TSP) (7, 8). Several studies aiming to discover replication inhibitors have bee reported. However, up-to-date, there are no reports on the successful control of infection (9).
HTLV-I Integrase (IN) is the enzyme responsible for the integration of reversely transcribed viral DNA (cDNA) into host cell DNA through two-step metal dependent reactions (10). In addition, IN plays an important role in HTLV-I genome replication, and it has been considered as one of the most promising targets for development of effective antiretroviral drugs.
Up-to-date, it has been reported that all known retroviral integrases present three domains: i) the zinc binding N-terminal (His12, His16, Cys40 and Cys43), ii) the catalytic core (Asp64, Asp116 and Glu152), and iii) the dsDNA-binding C-terminal domain (11, 12). There are available IN structural information on human immunodeficiency virus type 1 (HIV-1) (13- 15), avian sarcoma virus (ASV) (16), prototype foamy virus (PFV) (17) and HIV-1 with orthologous genes close to Lentivirus and Alpharetrovirus (18-20). Additionally, the high similarity of the tridimensional structure among all solved retroviral INs allows determining the structure of unsolved IN through homology modeling (21, 22).
Dicaffeoylquinic Acids (DCQAs) have been previously reported as potent and irreversible inhibitors of HIV-1 (2225). DCQAs have low cytotoxicity, present low molecular weight, have drug-like properties and are considered as promising inhibitors towards HIV-1 and HTLV-I integrases (23, 24). Despite the fact that its potent inhibitory activity is known, the underpinnings behind the inhibition mechanism remain to be elucidated.
Molecular Docking (MD) is a computational method capable of predicting the most probable topology during the formation of protein-ligand complexes (26) along with quantitative measurements of the affinity such as Binding Free Energy (BFE). This tool has been widely used to evaluate the potential of ligands as competitors which can abolish the infections (27, 28). MD coupled with other calculations, such as BFE (29), enables insights into biological systems and helps a more guided drug design.
In the present study, we evaluate the 3-5-DCQA interaction (IUPAC name: (S)-2-((1R,3R,4R,5R)-3,5-bis((E)-3-(3,4-dihydroxyphenyl)acryloyloxy)-4hydroxycyclohexyl)-2-hydroxyacetic acid) with the catalytic domain of Human T-Lymphotropic (HTLV-I) type I Integrase in the presence of Mg2+ ions. In this sense, a homology model of HTLV-I IN was proposed and docking analysis were performed between HTLV-I IN and 3,5-DCQA, considering the magnesium (Mg2+) ions influence. This modeling corroborated that the presence of one coordinated Mg2+ ion decreases the binding free energy between 3,5-DCQA and HTLV-I IN. Thus, we propose 3,5-DCQA as a pharmacophore for HTLV-I integration process inhibition.
Materials and methods
HTLV-I Integrase homology modeling
Simple Modular Architecture Research Tool (SMART) software (http://smart.embl-heidelberg.de/) was used to identify the catalytic site. HTLV-I IN core domain was located between amino acids 492 and 631, confirmed from information of Pfam Data Base (http://pfam.sanger.ac.uk/) (30). HTLV-1 Pol amino acids sequence (accession number BAH85786), available at National Center for Biotechnology Information, NCBI (http://www.ncbi.nlm.nih.gov/), was the basis for determining the catalytic domain of HTLV-I IN. Protein Data Bank (PDB) Non Redundant Database searches using matrix similarity BLOSUM62 with Basic Local Alignment Search Tool (BLAST) software (http://blast.ncbi.nlm.nih.gov/Blast.cgi) were used to obtain the homology template for HTLV-I IN. The catalytic domain of Avian Sarcoma Virus (ASV) Integrase (PDB: 1VSD) (16) showed the highest homology score.
The homology model was constructed using Prime software (Schrodinger, Inc). The HTLV-I IN secondary structure was determined by alignment of its core domain and ASV template sequences by means of Prime-Secondary Structure Prediction Module. The rotamers of the conserved amino acids were maintained during model building. Next, the side chains were optimized and the residues not derived from the template were minimized. Polak-Ribiere conjugate gradient, OPLS-2005 force field and GB/SA with implicit water-like solvent algorithms were used to optimize loops and complex minimization, using MacroModel (Schrodinger, Inc).
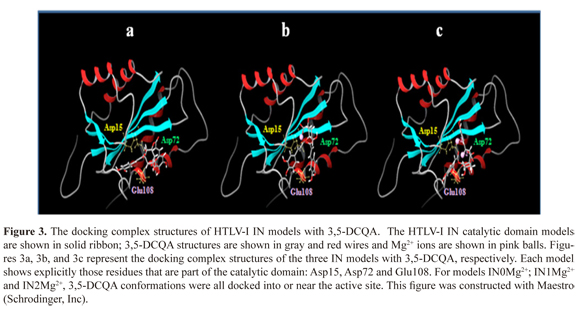
Three HTLV-I IN catalytic domain models, each one with different Mg2+number and position, were constructed: the first one without Mg2+ ions (IN0Mg2+); the second one with one Mg2+ ion coordinated at Asp15 and Asp72 (IN1Mg2+) and the third one with two Mg2+ ions, one chelated at Asp15-Asp72 and the other one at Asp72-Glu108 (IN2Mg2+). The Asp 15, Asp72 and Glu108 are highly conserved amino acids of the catalytic domain of IN for HTLV-1, being homologous to Asp64, Asp116 and Glu152 of IN for HIV-1.
The coordinates to incorporate the Mg2+ ions into the homology model of HTLV-I (models two and three), were obtained from 1QS4 (13) and 1VSH (16) crystal structure available in the Protein Data Bank (www.rcsb.org). Those models were achieved by modifying the PDB HTVL-1 model IN format and imported to Maestro file formats (Schrodinger, Inc) (31). Hydrogen atoms were added to IN with Protein Preparation Wizard workflow in Maestro (Schrodinger, Inc) and minimized using OPLS-2005 force field.
3,5-Dicaffeoylquinic acid modeling
The structure of 3,5-Dicaffeoylquinic Acid was constructed using the fragment dictionary of Maestro software 9.0 (Schrodinger, Inc). The ionization states were predicted with Epik (Application of MacroModel, Schrodinger, Inc) using water as solvent at a pH = 7.0±2.0. ConfGen program (Schrodinger, Inc) was used to identify the most stable ligand conformation with full mode 0.5 A as RMS and 120 kcal /mol energy window in order to eliminate redundant conformations. Finally, the structure was optimized with Jaguar software (Schrodinger, Inc) using Density Functional Theory method, with 6-31G** basis set.
Molecular docking
Glide 5.5 software (Schrodinger, Inc) was used for docking studies. To relax the ligand coupling at protein site, Glide uses a series of hierarchical filters to search possible locations of the receptor in the active site. The first filter localizes the ligand centre at various grid positions of a 1 Å grid and also rotates it around the three Euler angles. At this stage, crude score values and geometrical filters remove those unlikely binding modes. The next filter stage involves a grid-based force field evaluation and refinement of docking solutions including torsional and rigid body movements of the ligand. OPLS-2005 force field was used for this purpose. A small number of surviving docking solutions can then be subjected to a Monte Carlo procedure to minimize the energy score. The final energy evaluation with GlideScore was carried out and a single best pose was generated as output for a particular ligand (32).
The GlideScore scoring function is based on ChemScore, but includes a steric-clash term, adds buried polar terms devised by Schrodinger to penalize electrostatic mismatches, and has modifications to others terms. GlideScore modifies and extends the ChemScore as follow:
GScore = 0.065*vdW + 0.130*Coul + Lipo + Hbond + Metal + BuryP + RotB +Site
Where: vdW= van der Waals energy; Coul=Coulomb energy; Lipo=lipophilic contact term; HBond=hydrogen-bonding term; Metal=metal binding term; BuryP= penalty for buried polar groups; RotB= penalty for freezing rotatable bonds; Site=polar interactions at the active site.
After preparing and minimizing the three models (IN0Mg2+, IN1Mg2+ and IN2Mg2+) and the 3,5-DCQA, we used the receptor grid generation application of Glide to construct the receptor-grid files. To soften the potential non-polar regions of each IN models we scaled van der Waals radius of the receptor atoms by 1.00 Å with a partial atomic charge of 0.25. The size outcome of grid box was 13 x 13 x 13 Å in each dimension, with coordinates: X=49.9592, Y=35.3464 and Z=60.5208 and centered at the active site delimited at residues Asp15, Asp72 and Glu108. To define the spherical region position that should be occupied by the ligand during docking, Mg2+ ions were used as constraints in the second and third models. The Mg2+ ions and the ligand were selected from workspace.
To perform the docking calculations, we used the "Extra Precision" Glide algorithm. XP GlideScore is a function that severely penalizes poses that violate established physical and chemistry principles, e.g. charged and strong polar groups forming H-bonds or adequately being exposed to the solvent (33). This algorithm is broadly used to minimize positive falses and is highly useful for studies in which the number of structures is limited and pursue the highest possible computational quality.
Re-docking
Re-docking procedure for the three integrases models were performed afterwards to obtain the GlideScore data. The procedure was carried out using the workflow QM-Polarized Ligand Docking by recalculating charges using quantum mechanics at the site. The best scores were used to recalculate the charges of 3,5-DCQA and also to perform the Re-docking by using Qsite and Jaguar (Schrodinger Inc). The 3,5-DCQA charges were obtained from the electrostatic potential energy surface which was evaluated from calculations in a single point using density functional theory (DFT) under 6-31G*/LACVP* basis set, B3LYP functional, Ultrafine SCF accuracy level parameters. Once the charges were calculated, a new docking was performed in XP mode, excluding those poses that were duplicated (Root Mean Square deviation value is less than 5 Å and the atomic displacement is less than 1.3 Å). The final pose was selected based on Coulomb-vdW energy.
Binding free energy calculations
In order to select which complex pose displays the best affinity, we evaluated the binding free energy (BFE). The best re-docking scoring pose was taken for BFE calculations. Energy difference mode framed in eMBrAcE software (Schrodinger Inc.) was used for calculating receptor, ligand and complex energies, and the energy change was evaluated based on the following equation:
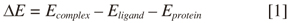
The values of free energy for the three complexes were subjected to energy minimization in implicit solvent (water) using OPLS-2005 force field with dielectric constant of one. PRCG method was used to calculate solvation energy of electrostatic component. A conjugate gradient minimization protocol with 10,000 maximum steps of iterations and convergence threshold of 0.05 was used to determine the minimum energy.
Results
Homology modeling
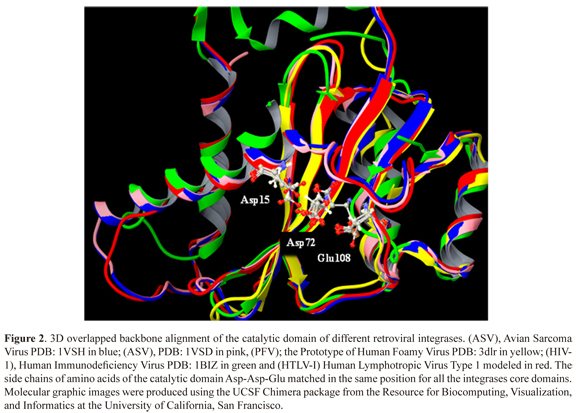
The HTLV-I Integrase model that was obtained by homology modeling (homology 44%, identity 34% and positivity 44%) (Figure 1), presented well structured regions of five beta sheets and four alpha helices. The well- structured regions displayed high similarity with other integrases that have been analyzed by X-Ray diffraction, which include the Avian Sarcoma Virus (ASV, pdb: 1VSD and pdb: 1VSH); Human Immunodeficiency Virus (HIV-1, pdb: 1BIZ) and Prototype of Human Foamy Virus (PFV, pdb: 3DLR) (Figure 2).
Docking and re-docking
Docking and re-docking were performed in order to analyze the main physicochemical and thermodynamically issues of the interaction of 3,5 DCQA with the catalytic domain of HTLV-I IN (Figure 3).
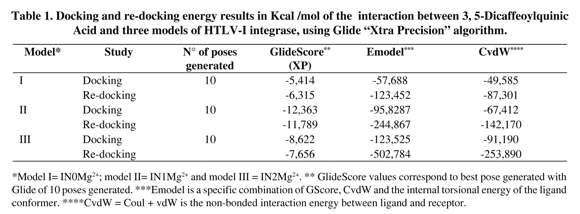
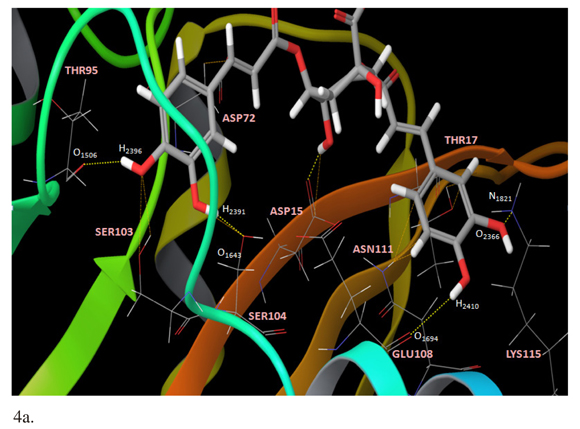
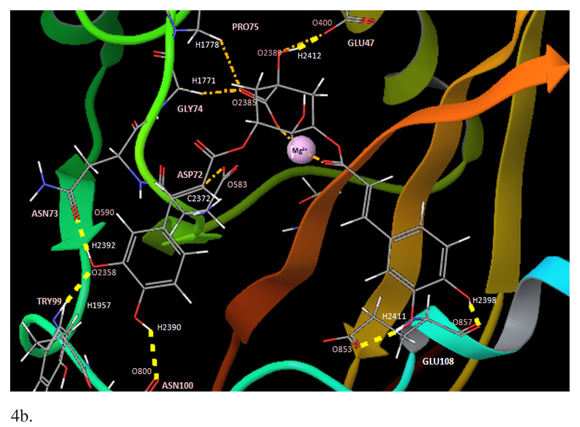
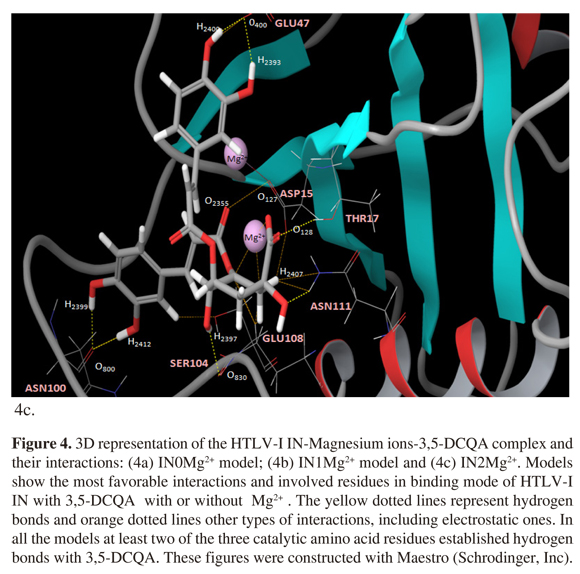
The presence of one Mg2+ ion (IN1Mg2+) showed the best affinity interaction characterized by a low score value in GlideScore (-12,363 kcal/mol). These results were corroborated with quantum mechanics calculations (Table 1). The complex 3,5-DCQA-HTLV-I IN interactions were analyzed in major dock score. Interestingly, we identified interactions between 3,5-DCQA and the functional domain amino acids (Asp15-Asp72-Glu108); for all the cases thirteen interactions for IN0Mg2+ were observed, four of them corresponding to hydrogen bonds (Figure 4a). Similarly, thirteen interactions were observed for IN1Mg2, with two electrostatic types between ligand, caffeic acid group and Mg2+ ion, and six hydrogen bonds (Figure 4b). In this model, Glu108 and Asp72 established H-bonds with 3,5-DCQA. Finally, IN2Mg2+ presented the highest number of interactions (23 in total); six H-bonds and six electrostatic bonds connected with Mg2+ ions. In this model, Asp15 and Glu108 bound through H-bonds to 3,5-DCQA (Figure 4c).

Binding free energy
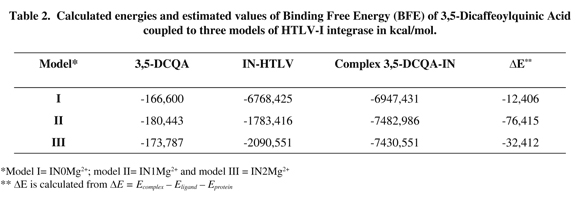
Binding free energy calculations showed that the presence of one Mg2+ ion presents the best affinity, confirming the docking and re-docking results (Table 2).
Discussion
In this in silico study we reported for the first time a homology model of the catalytic domain of integrase (IN) for HTLV-I. Comparison of our homology model with some crystallographic structures of IN for other viruses, such as: HIV-1 (1BIZ), VSA (1VSD, 1VSH) and PFV (3DLR) showed a high similarity structure, especially in the positions of the amino acid of the catalytic site, Asp15, Asp72 and Glu108, suggesting that these amino acids are very important for the enzymatic function of IN. Additionally, these results support our homology model of IN for HTLV-1.
We found that the presence of Mg2+ ions as protein cofactor in vivo could affect the binding between the IN of HTLV-1 and the chemical compound 3,5-DCQA as well. A single Mg2+ ion can stabilize the complex between IN and 3,5-DCQA, decreasing the interaction energy and showing a more specific binding. The same results were obtained using different mathematical models simulations, such as: Docking, Re-docking and BFE.
It is important to notice that a single Mg2+ ion could increase the binding stability between 3,5-DCQA and IN of HTLV-1, the 3,5 - DCQA itself can bind within or near to the catalytic domain of IN and set up interactions with at least two of the three amino acids of the catalytic domain (Asp15, Asp72 and Glu108). This result suggests that 3,5-DCQA is an important molecule with a high pharmaceutical potential against IN of HTLV-1, and therefore, further experimental studies are needed.
Regarding the interactions between IN, Mg2+ ions and 3,5-DCQA generated in each model, the higher affinity was observed in the interaction between IN1Mg2+ and 3,5-DCQA. However, the interactions in model IN2Mg2+ were more frequent (24 interactions) compared with the interactions in IN1Mg2+ and IN0Mg2+ models (13 interactions each). One possible explanation for this result could be that the interactions in IN1Mg2 + are stronger, such as hydrogen bonds, in comparison with the other two models, where the energy of the system increases and improves the affinity of interaction. This aspect was evident for the GlideScore value obtained in this study.
Similar studies have been performed in the IN of HIV-1 in order to evaluate the influence of Mg2+ in molecules with a potential inhibitor to IN of HIV-1 via binding free energy calculations. Wang and coworkers (37) reported in an in silico study the influence of Mg2+ ions on the binding between IN of HIV-1 and the inhibitor molecule Thiazolothiazepine (2H-benzo(f)thiazolo(2,3-c)(1,4)thiazepine-5,11(3H, 11aH)-dione) using computer simulation and methodologies such as docking and molecular dynamics. Results showed that the presence of Mg2+ ions could effectively avoid or influence the binding between them. Also, that the presence of a single Mg2+ ion coordinated by Asp 64 and Asp116 decreases the docking energy and determines a more specific binding. However, the presence of a second Mg2+ ion coordinated by Asp64 and Glu152 is disadvantageous for the interaction, since it increases the range of energy and decreases the affinity of the coupling (34). Our results are in agreement with the results obtained by Wang et al. (34) for the interaction between 3,5-DCQA and IN of HTLV-1, although the ligand molecules showed a different structure.
Other authors have suggestes that there is not enough countercharge in the catalytic domaun of the enzyme to allow simultaneous coupling of two divalent cations in adjacent sites (35, 36). Additionally, it has been proposed that although there are two Mg2+ ions in the catalytic site, in the persence of viral DNA only one would be available to interact with one incoming inhibitor molecule, because the second ion may be largely shielded, while the first ion could be much more exposed (37).
Based on experimental evidence of the crystal structure of the IN of Avian Sarcoma Virus (ASV), it has been suggested that there could be two Mg2+ ions interacting in the catalytic domain of IN for HIV-1 (16). However, so far only the presence of one Mg2+ ion of IN for HIV-1 has been experimentally characterized. Unlike what was initially expected, our studies showed greater affinity between the IN of HTLV-1 and 3,5-DCQA when one Mg2+ ion is present. The experimental observation supports our proposed predictive results of IN for HTLV-1.
Conclusion
We calculated the binding model between IN of HTLV-1 and the chemical compound 3,5-DCQA using molecular modeling, such as Docking, Re-docking and Binding Free Energy (BFE). The results confirmed that the presence of Mg2+ ions could affect the binding between 3,5-DCQA and the IN enzyme of HTLV-1. Also, we found that the presence of a single Mg2+ ion in the interface ligand-enzyme favors the interaction of the complex. Additionally, it was determined that regardless of the presence of Mg2+ ions important for catalytic activity of the enzyme, 3,5-DCQA established interactions with any of the amino acids that are highly conserved in the IN enzyme catalytic site of HTLV-1. Therefore, we propose that the 3,5-DCQA is a molecule with an anti-integrase pharmaceutical potential.
Acknowledgments
We want to extend our appreciation to: Dr. Andrés González Barrios (Universidad de los Andes, Colombia) and Dr. Mihaela Delcea (University of Greifswald, Germany) for critical reading of the manuscript.
Financial support
This work was partially supported by grants of COLCIENCIAS (Young Researchers Program) and the Vice-Rectorate of the Pontificia Universidad Javeriana (PUJ 001648).
Conflict of interest
The authors declare that there are no conflicts of interest.
References
1. Edlinch RF. Arnette JA. Williams FM. Global epidemic of human T-cell lymphotropic virus type-I (HTLV-I): an update. Journal of Long-Term Effects of Medical Implants 2003 13: 127-140. [ Links ]
2. Michaelsson J. Barbosa HM. Jordan KA. Chapman JM. Brunialti MK. Neto WK. Nuui Y. Sabino EC. Chieia MA. Souza A. Oliveira B. Nixon DF. Kallas EG. The frequency of CD127 low expressing CD4+CD25 high T regulatory cells is inversely correlated with human T lymphotrophic virus type-1 (HTLV-I) proviral load in HTLV-I-infection and HTLV-I-associated myelopathy/tropical spastic paraparesis. BMC Immunology. 2008 9(41): 1-12. [ Links ]
3. Proietti FA. Carneiro-Proietti AB. HTLV in the Americas. Pan American Journal of Public Health. 2006 19: 7-8. [ Links ]
4. Edlich R. Arnette J. Williams FM. Global Epidemic of Human T-Cell Lymphotropic Virus Type-1 (HTLV-I). Journal of Emergency Medicine. 2000 18: 109-119. [ Links ]
5. Proietti FA. Carneiro-Proietti AB. Catalan-Soares BC. Murphy EL. Global epidemiology of HTLV-I infection and associated diseases. Oncogene. 2005 24: 6058-6068. [ Links ]
6. Yoshida M. Seiki M. Yamaguchi K. Takatsuki K. Monoclonal integration of human T-cell leukemia provirus in all primary tumors of adult T-cell leukemia suggests causative role of human T-cell leukemia virus in the disease. Proceedings of the National Academy of Science of the United States of America. 1984 81: 2534-2537. [ Links ]
7. Gessain A. Barin F. Vernant JC. Maurs L. Gout O. Calender A. De G. Antibodies to human T-lymphotropic virus type-I in patients with tropical spastic paraparesis. Lancet. 1985 2: 407-410. [ Links ]
8. Osame M. Janssen R. Kubota H. Nishitani H. Igata A. Nagataki S. Mori M. Goto I. Shimabukuro H. Khabbaz R. et al. Nationwide survey of HTLV-I-associated myelopathy in Japan: association with blood transfusion. Annals of Neurology. 1990 28: 50-56. [ Links ]
9. Balestrieri E. Matteucci M. Ascolani A. Piperno A. Romeo R. Romeo G. Chiacchio U. Mastino A. Macchi B. Effect of Phosphonated Carbocyclic 2'-Oxa-3'-Aza-Nucleoside on Human T-Cell Leukemia Virus Type 1 Infection In Vitro. Antimicrobial Agents and Chemotherapy. 2008 52: 54-64. [ Links ]
10. Diamond TL. Bushman FD. Role of metal ions in catalysis by HIV integrase analyzed using a quantitative PCR disintegration assay. Nucleic Acids Research. 2006 34: 6116-6125. [ Links ]
11. Dyda F. Hickman AB. Jenkins TM. Engelman A. Craige R. Davies DR. Crystal Structure of the Catalytic Domain of HIV-1 Integrase: Similarity to Other Polynucleotidyl Transferases. Science. 1994 266: 1981-1986. [ Links ]
12. Healy EF. Sanders J. King PJ. Robinson WE. A docking study of L-Chicoric acid with HIV-1 integrase. Journal of Molecular Graphics and Modelling. 2009 27: 584-589. [ Links ]
13. Goldgur Y. Dyda F. Hickman AB. Jenkins TM. Craigie R. Davies DR. Three new structures of the core domain of HIV-1 integrase: An active site that binds magnesium. Proceedings of the National Academy of Science of the United States of America. 1998 95: 9150-9154. [ Links ]
14. Goldgur Y. Craigie R. Cohen GH. Fujiwara T. Yoshinaga T. Fujishita T. Sugimoto H. Endo T. Murai H. Davies DR. Structure of the HIV-1 integrase catalytic domain complexed with an inhibitor: A platform for antiviral drug design. Proceedings of the National Academy of Science of the United States of America. 1999 96: 13040-13043. [ Links ]
15. Molteni V. Greenwald J. Rhodes D. Hwang Y Kwiatkowski W. Bushman FD. Siegel JS. Choe S. Identification of a small molecule binding site at the dimer interface of the HIV integrase catalytic domain. Acta Crystallographica Section D. 2001 57: 536-544. [ Links ]
16. Bujacz G. Alexandratos J. Wlodawer A. Binding of Different Divalent Cations to the active Site of Avian Sarcome Virus Integrase and Their Effects on Enzymatic Activity. Journal of Biological Chemistry. 1997 272: 18161-18168. [ Links ]
17. Volkov E. Gupta SS. Hare S. Helander A. Roversi P. McClure M. Cherepanov P. Functional and structural characterization of the integrase from the prototype foamy virus. Nucleic Acids Research. 2009 37: 243-255. [ Links ]
18. Hare S. Di Nunzio F. Labeja A. Wang J. Engelman A. Cherepanov P. Structural Basis for Functional Tetramerization of Lentiviral Integrase. PLoS Pathogen. 2009 5(7): 1-15. [ Links ]
19. Yang ZN. Mueser T. Bushman FD. Hyde CC. Crystal Structure of an Active Two-domain Derivate of Rous Sarcoma Virus Integrase. Journal of Molecular Biolog. 2000 296: 535-548. [ Links ]
20. Lubkowski J. Dauter Z. Yang F. Alexandratos J. Merkel G. Skalka AM. Wlodawer A. Atomic Resolution Structures of the Core Domain of Avian Sarcome Virus Integrase and Its D64N Mutant. Biochemistry. 1999 38: 13512-13522. [ Links ]
21. Tsai S. Molecular Modeling: Protein Modeling. In: An Introduction to Computational Biochemistry.First edition. Wiley-Liss Inc Publisher New York 2002 pp 315-341. [ Links ]
22. Robinson WE. Reinecke MG. Abdel-Malek S. Jia Q. Chow SA. Inhibitors of HIV-1 replication that inhibit HIV integrase. Proceedings of the National Academy of Science of the United States of America. 1996 93: 6326-6331. [ Links ]
23. Zhu K. Cordeiro ML. Atienza J. Robinson EJr. Chow S. Irreversible Inhibition of Human Immunodeficiency Virus Type 1 Integrase by Dicaffeoylquinic Acids. Journal of Virology. 1999 73: 3309-3316. [ Links ]
24. McDougall B. King PJ. Wu B. Hostomsky Z. Reinecke M. Robinson EJr. Dicaffeoylquinic and Dicaffeoyltartaric Acids Are Selective Inhibitors of Human Immunodeficiency Virus Type 1 Integrase. Antimicrobial Agents and Chemotherapy. 1998 42: 140-146. [ Links ]
25. Tamura H. Akioka T. Ueno K. Chujyo T. Okazaki K. King PJ. Robinson EJr. Anti-human immunodeficiency virus activity of 3 4 5-tricaffeoyilquinic acid in cultured cells of lettuce leaves. Molecular Nutrition & Food Research. 2006 50: 396-400. [ Links ]
26. Taylor RD. Jewsbury PJ. Essex JW. A review of proteinsmall molecule docking methods. Journal of Computer-Aided Molecular Design. 2002 16: 151-166. [ Links ]
27. Thomsen R. Flexible ligand docking using evolutionary algorithms: investigating the effects of variation operators and local search hybrids. ByoSystems. 2003 72: 57-73. [ Links ]
28. Kitchen DB. Decornez H. Furr JR. Bajorath J. Docking and Scoring in Virtual Screening for Drugs Discovery: Methods and Applications. Nature Reviews/Drug Discovery. 2004 3: 935-949. [ Links ]
29. Brandsdal BO. Österberg F. Almlöf M. Feierberg I. Luzhkov VB. Åqvist J. Free Energy Calculations and Ligand Binding. Advances in Protein Chemistry. 2003 66: 123-158. [ Links ]
30. Finn RD. Tate J. Mistry J. Coggill PC. Sammut SJ. Hotz HR. Ceric G. Forslund K. Eddy SR. Sonnhammer EL. Bateman A. The Pfam protein families database. Nucleic Acids Research. 2008 36: D281-288. [ Links ]
31. Maestro version 9.0 2009. Schödinger LLC New York NY [ Links ]
32. Halgren TA. Murphy RB. Friesner RA. Beard HS. Frye LL. Pollard WT. Banks JL. Glide: a new approach for rapid accurate docking and scoring. 2. Enrichment factors in database screenin. Journal of Medicinal Chemistry. 2004 47: 1750-1759. [ Links ]
33. Friesner RA. Murphy RB. Repasky MP. Frye LL. Greenwood JR. Halgren TA. Sanschagrin PC. Mainz DT. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. Journal of Medicinal Chemistry. 2006 49: 6177-6196. [ Links ]
34. Wang L. Influence of Mg2+ on the binding modes of HIV-1 integrase with thiazolothiazepine inhibitor studied by molecular simulation. Computers in Biology and Medicine. 2009 39: 355-360. [ Links ]
35. Grobler JA. Stillmock K. Hu B. Witmer M. Felock P. Espeseth AS. Wolfe A. Egbertson M. Bourgeois M. Melamed J. Wai JS. Young S. Vacca J. Hazuda DJ. Diketo acid inhibitor mechanism and HIV-1 integrase: Implications for metal binding in the active site of phosphotransferase enzymes. Proceedings of the National Academy of Science of the United States of America. 2002 99: 6661-6666. [ Links ]
36. Chen X. Tsiang M. Yu F. Hung M. Jones GS Zeynalzadegan A Qi X. Jin H. Kim CU. Swammathan S. Chen JM. Modeling Analysis and Validation of a Novel HIV Integrase Structure Provide Insights into the Binding Modes of Potent Integrase Inhibitors. Journal of Molecular Biology. 2008 380: 504-519. [ Links ]
37. Wang L. Liu CL. Chen WZ. Wang CX. Constructing HIV-1 integrase tetramer and exploring influences of metal ions on forming integrase-DNA complex. Biochemical and Biophysical Research Communications. 2005 337: 313-319. [ Links ]

