











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroduction
Soil is considered a unique environment containing a considerable diversity of organisms, of which only 1 % can be cultivated in the laboratory (Daniel 2005; van Elsas et al. 2008). However, a promising tool for understanding microbial communities in different ecosystems is metagenomics. This approach facilitates the study of entire genomes directly from the environmental samples (Handelsman 2004; Daniel 2005; Bonilla-Rosso et al. 2008; Angelov et al. 2009; Fu et al. 2011; Gomes et al. 2013). Brazilian Atlantic Forest is considered to be one of the most
important biosphere reserves and it exhibits a high biodiversity. The United Nations Educational, Scientific and Cultural Organization has declared this area to be a “World Hotspot” (http://wwwpmma.etc.br), an ecosystem that plays an important role in the formation of microhabitats; the environmental pressure on microorganisms encourages diversification (Chenu & Stotzky 2002).
The isolation of secondary metabolites produced by microorganisms historically has been effective for the identification of new compounds with bioactive properties. These compounds supply a large number of molecules that are currently in commercial use such as antibiotics, antifUngals, immunosuppressants, and antitumorals (Hopwood, 2004). These secondary metabolites include a group of molecules called polyketides, which are a family of structurally diverse compounds. The richness and diversity of the secondary metabolism in this group add valuable bioactive molecules with pharmaceutical activity. Among the large number of industrial uses of polyketide drugs, cephalosporins, rifamycins, tetracyclines, erythromycin, different p lactams and anthracyclines stand out; many polyketides have potential for the development of commercial drugs. Despite their diversity, they share the same biosynthetic pathway steps carried out by the catalysis of enzymes called polyketide synthases (PKS) (Hopwood 1993; Hutchinson & Fujii 1995; Staunton & Weissman 2001; Hopwood 2004). The highly modular nature of PKS biosynthetic systems provides a template for the discovery of a wide variety of gene clusters that give rise to a diverse chemical repertoire, including many of the most clinically useful microbial metabolites (Cragg & Newman 2013).
In this study we employed a metagenomic approach to analyze the biosynthetic diversity of the PKS and ketosynthase (KS) domains in a metagenomic library from an Atlantic Forest soil sample.
Materials and methods
Soil collection and DNA isolation
Soil samples were collected from the Atlantic Forest located in Ilhabela São Paulo State, Brazil 23° 51' 7" S 45° 20' 38" W in a delimitated space of 2 m2 at 30 cm deep, according to the methodology proposed by (Berry et al. 2003). The soil was calcaric. DNA was extracted from the soil using an UltraClean® Soil DNA Isolation Kit (Mobio Inc., Carlsbad, CA, USA). The manufacturer’s instructions were modified to obtain large DNA fragments, as follow: DNA was filtered twice through column purification. For a third cleaning step a new column was used, following precipitation with a solution of guanidine 10 mM. DNA was washed once with ethanol 95 % and finally incubated overnight at -20 °C.
Bacterial artificial chromosome library construction
A metagenomic library was constructed using the pJW360 vector (Wild et al. 2002). The purified metagenomic DNA was randomly sheared and size-selected around 50Kb before being cut with Sal I (New England Biolabs Inc, Ipswich, Massachusetts) and ligated into the pJW360 vector. DNA was cloned into an Escherichia coli DH10B strain and plated onto Luria-Bertani containing 12.5 mg/mL chloramphenicol for the selection of the recombinant clones. To induce the production of higher bacterial
artificial chromosome (BAC) copy numbers, arabinose was added (0.01 % v/v) to the medium with the targeted clones. The recombinant Escherichia coli clones were picked in polypropylene plates; a total of 5, 000 clones (library size) were obtained and tested.
Library screening
BAC DNA was extracted using a GeneJET Plasmid Miniprep Kit (Life Technologies, Carlsbad, California, USA). BAC DNA served as a template in a PCR reaction using degenerate primers targeting conserved regions in PKS. The amplification for PKS type I was carried out by multiplex PCR as follow: the first PKSIsetl: sense, 5-CCSCAGSAGCGCSTSTTSCTSGA-3, and antisense, 5-GTSCCSGTSCCGTGSGTSTCSA-3; PKSIset2: sense, 5-CCSCAGSAGCGC STSCTSCTSGA-3, and antisense, 5-GTSCCSGTSCCGTGSGCCTCSA-3 (Courtois et al. 2003). The PCR parameters were 20 s of denaturation at 95 °C followed by 30 cycles (95 °C for 60 s, annealing at 62 °C for 60 s, extension at 72 °C for 60 s and then 300 s at 72 °C). Amplification for PKS type II was carried out by classic PCR, as follow: primer sense 5'- TSGCSTGCTTCGAYGCSATC-3' and antisense 3'- TCGCCBAAGCCGCCNAAGGT-5' (Metsa-Ketela et al., 1999) were used with the parameters 60 s of denaturation at 95 °C followed by 30 cycles (95 °C for 60 s, annealing at 58 °C for 60 s, extension at 72 °C for 60 s, and then 510 s at 72 °C). To confirm PKS-positive clones, the PCR product was analysed on 1 % agarose gel- 1X TBE run at 3V/cm2 for 120 min; followed by sequencing of the PCR products (50 ng/pL) using the screening primers with an Applied Biosystems 3730 instrument.
Bioinformatics analysis of BAC-derived positive clones
The sequence data were proofread using Chromas version 1.45 (Technelysium Pty Ltd., Australia). The generated DNA sequences were compared by using a database of the National Center for Biotechnology Information (NCBI - GenBank). Putative gene functions were identified using the BLASTx and BLASTp algorithms. Furthermore, we studied the sequence protein homology using the server Phyre 2 (Kelley & Sternberg 2009). Phylogenetic analysis was carried out using the CLUSTAL-W program in MEGA 6.06 (Tamura et al. 2013). The tree topologies were evaluated by bootstrap analyses based on 1,000 replicates, and phylogenetic trees were generated using the neighbour-joining (NJ method (Saitou & Nei 1987).
Results and Discussion
PCR-based metagenomic library screening for PKS genes
The recombinant BAC DNA from 5,000 clones from the soil metagenomic library was used as template for PCR. A positive PCR result for the PKS domains was recovered from 2 clones (Table 1). Compared with other metagenomic libraries constructed using large-insert fosmids and BAC vectors, this one includes a low number of positive clones. Courtois et al. (2003) reported 11 different KS domain sequences detected in a 5,000 clone library. Metagenomic libraries with more than 10,000 clones did not have numbers better than 34 positive clones (Ginolhac et al. 2004; van Elsas et al. 2008; Parsley et al. 2011). Other approaches, such as cloning vectors like BACs exhibited considerable differences compared with fosmids in metagenomic libraries for PKS screenings (Parsley et al. 2011).
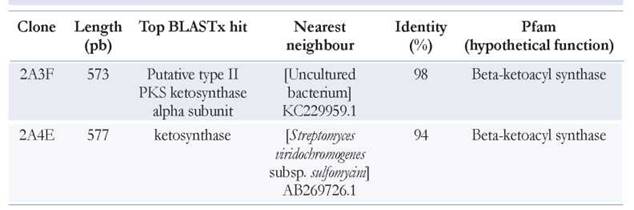
Bioinformatics predicts protein function
An important tool to enhance the power of the metagenomic method is bioinformatic analysis, which supports the identification of new genes for several medical and industrial molecule pathways (Gomes et al. 2013). The protein sequences exhibited identities of up to 90 % with published sequences according to BLAST analysis, and the function protein was confirmed in the Pfam database (Table 1).
Interestingly the clone 2A4E can be grouped in the Streptomycetaceae family, and the closest neighbour to S. viridochromogenes (Figure 1); this bacteria produce streptazolin, a neutral tricyclic compound known for antibiotic and antifungal activities. A general retrosynthetic analysis for streptazolin analogues showed a unique structural feature as well as its promising biological activity profile (Nomura & Mukai 2004). We consider that enzymes involved in the synthesis of streptazolin-related natural products from metagenomic samples can provide structural features for developing antifungal molecules.
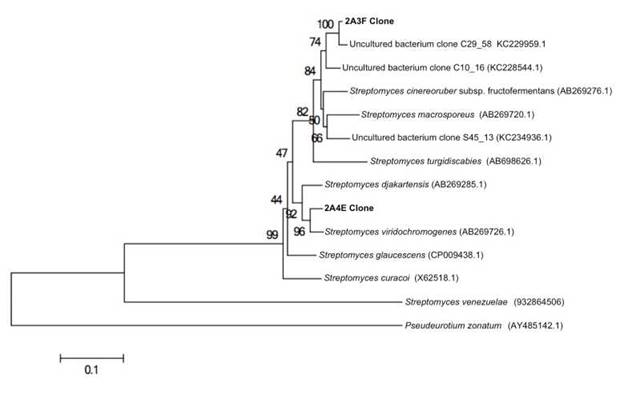
Fig. 1 Phylogenetic analysis of the ketosynthase genes. The NJ tree is based on cultivable strains and metagenomic clones. Pseudeuritium zonatum (AY 485142.1) was used as the outgroup. Bootstrap analysis (performed 1,000 times). Scale bar represents 0.1 amino acid substitutions per site.
The two sequences exhibited a functional beta-ketoacyl synthase. Additionally, the Server Phyre 2.0 revealed a 73 % identity, a confidence of 100 %, and the same catalytic residues with a ketosynthase/chain length factor (Id: c1tqyC_, Access Fri Jul 3 22:16:53). This enzyme catalyses the condensation of malonyl-ACP (Zhang et al. 2001), and it is involved in the biosynthesis of polyketide aromatic antibiotics (Staunton & Weissman 2001). This family includes medermycin, kalafungin, granaticin, and actinorhodin, a well-known compound used as a model for studying the biosynthetic mechanisms of aromatic polyketide antibiotics. These compounds possess strong antibacterial activity and have a distinctive structure composed of a benzene, a quinone, and a stereospecific pyran ring (Lu et al. 2015).
Analogous approaches for screening polyketides in a soil sample from Jaboticabal, State of Sao Paulo, were carried out (Gomes et al. 2013). These authors found a PKS module that exhibited homology with myxobacteria PKS type I (36 % identity and a 50 % BLASTp similarity with Chondromyces crocatus CndA protein, its closest neighbour). Gomes et al. (2013) suggested that additional studies devoted to the expression and inactivation of these genes are necessary to prove the role of the discovered genes in polyketide biosynthesis. However, their study demonstrated that metagenomic DNA represents a promising source for screening new genes that can be used in combinatorial biosynthesis experiments aimed to the production of new molecules.
In this study, the focus was on an enzyme directly involved in the biosynthesis of polyketide type II compounds. The analyses of alpha-ketoacyl synthase (KSA) subunits suggest an elongation function for the biosynthesis of these kinds of bioactive molecules; the differences in the sequence may reflect an unknown molecule. Several combinations of PKS genes from different origins have been expressed in suitable hosts to generate a wide variety of compounds exhibiting novel structural properties not found in natural products produced by bacterial strains (Metsa-Ketela et al., 1999). The compatibility among PKS from different pathways is common, making them a promising material for combinatorial biosynthesis and molecular engineering (Hopwood, 2004).
We suggest that future studies employ combinatorial biosynthesis to introduce metagenomic genes into secondary metabolite producer microorganisms, capable of expressing the new genetic information into new molecules. For PKS systems, some heterologous hosts are available such as Streptomyces litidans, S. albus, and S. coelicobr. This method can result in the production of polyketide structures due to the expression level control of these metabolites (Baltz 1998; Lombo et al. 2009; Medema et al. 2011).
We emphasize the importance of inferring the catalytic functions of putative enzymes via bioinformatics tools in metagenomic approaches. These strategies are validated through heterologous host expression, however it is suggested “shotgun” sequencing before constructing metagenomic libraries because the results can confer a general idea about the genes involved in secondary metabolism.
Considering the low number of sequences found in this work, they were compared with previous estimates of KS domain in different metagenomic libraries. The PKS- containing clone frequency was between 0.2 and 0.4 % from soil metagenomic libraries (Ginolhac et al. 2004; Parsley et al. 2011; Gomes et al. 2013); in this work it was of 0.06 %. The advantage of the use of BAC vectors was the average insert size compared with fosmid libraries; however, this study does not report a high frequency of KS-positive clones comparing fosmid libraries. We can suggest for future constructions to increase the number of clones, selection of new vectors, different approaches for screening, and massive sequencing studies before constructing metagenomic libraries to confirm the PKS diversity in environmental samples.
Regarding the KS domains found in this work, they are phylogenetically and probably functionally diverse, including some expected to be involved in cyclic and polyketide synthesis, as well as hybrid polyketide/non-ribosomal peptide chain elongation.
Conclusions
Few studies show the microbial biodiversity of the Atlantic Forest, as well as its biotechnological potential and even fewer show the antimicrobial prospective produced by bacteria present in soil. The methodology developed in this study will assist future studies for obtaining new PKS genes, such as probes, for screening metagenomic libraries in the search for new biosynthetic pathways.
This approach allowed a way for generating new compounds by combinatorial biosynthesis. The results show ketosynthase genes involved in the synthesis of polyketides type II. Those genes can be used for cloning in Streptomyces host to generate or modify compounds with new pharmacological activities.

