



![Synthesis, characterization and theoretical calculations of Cu(I) complex of trithiocyanuric acid [Cu(ttc)3]](/img/en/prev.gif)







 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroduction
Trypanosoma cruzi is the etiological agent of Chagas disease, an illness recognized for the World Health Organization [WHO] as one of the today seventeen-neglected tropical diseases [NTDs]. Indeed, this disease is a worldwide public health problem, with a current prevalence of 6 to 7 million of infected people, of which 0.7 to 1.2 million are in Colombia [1,2].
Like other medically important trypanosomatids, T. cruzi has a complex life cycle that must alternate between two types of hosts: insect triatomines and mammals, including humans [3]. To accomplish it, parasites must face and adapt to their hosts’ environments through a fine and delicate regulation of their gene expression. In these organisms, gene expression is largely regulated at the posttranscriptional level because of their special gene arrangement, in which large clusters comprising up to hundreds of genes and having the same transcriptional orientation, are constitutively expressed as polycistronic RNA precursors [4-7]. These polycistronic RNAs are ultimately processed into individual molecules through the addition of a Spliced Leader [SL], a common miniexon sequence present in all the mature mRNAs of trypanosomatids, to the 5’ end and a poly [A] tail to the 3’ end, processes known as trans-splicing and polyadenylation, respectively [8]. In this way, the mechanisms that govern mRNA expression basically operate at the maturation, transport, stability and translation steps through the recognition of RNA motifs, mainly located on the untranslated regions [UTR] of mRNAs, by RNA Binding Proteins [RBP]; both types of molecules, mRNAs and RBP, conform ribonucleoprotein complexes [RNP], which define the fate of the mRNA molecules [4-6,9,10].
In trypanosomatids, it has also been demonstrated the existence of alternative trans-splicing in which two or more mature transcripts are generated from the same gene by the use of different trans-splicing acceptor sites. The relevance of alternative trans-splicing is well illustrated in regards with the functional expression of the LYT1 protein, a virulence factor of T. cruzi [11,12]. This protein was uncovered in a pioneer study by Dr Andrews’ group in which it was described the existence in T. cruzi of a secreted protein, immunologically related to the C9 component of the membrane attack complex of complement, that possesses membrane pore-forming activity at low pH [13]. Given this activity, it was postulated that this protein would be mediating the escape of T. cruzi from the phagosome into the cytosol after cell invasion. Subsequently, Manning-Cela etal. [14], in an outstanding work, undertook the search for the coding gene of this virulence factor by immunoscreening of a T. cruzi cDNA expression library using antibodies against the C9 component. As a result, the LYT1 gene was identified and cloned. These authors, furthermore, demonstrated the cytolytic activity of the LYT1 protein by transfecting Schizosaccharomyces pombe with the LYT1 gene and analyzing its hemolytic effect [14]. Accordingly, deletion of LYT1 resulted in attenuation of T. cruzi infection capacity; however, unexpectedly, the LYT1-deficient epimastigotes transformed into metacyclic trypomastigotes more rapidly than wild-type parasites. Thus, a double function was suggested for LYT1: (i) a pore-forming activity relevant for intracellular survival, and (ii) a regulatory role during stage transition. Interestingly, in a subsequent article [15], these authors suggested the existence of two LYT1 isoforms, each one associated with a distinct functional role. These isoforms are originated by alternative trans-splicing events that generate different LYT1 mRNAs, which are translated in two different proteins: a full-length protein and an N-terminal truncated one. New articles by this group showed that the shorter protein (lacking 28 N-terminal amino acids, named kLYTl) is located at two spots in the mitochondrial kinetoflagellar zone and its presence was consistent with a role in the parasite development, whereas the larger product, mLYTl, localizes on the plasma membrane and would be responsible for its pore-forming function [16,17].
In terms of the LYT1 transcripts expression in CL Brener, a Discrete Typing Unit [DTU] VI strain; it is known that the relative abundance of the transcripts varies, within the parasite life cycle, from one stage to another. Indeed, the transcripts that give rise to the mLYTl isoform represent 65 % of the mRNAs derived from the LYT1 gene in trypomastigotes and amastigotes, whereas only 35 % corresponds to the kLYTl transcript in these two stages. In contrast, in the epimastigote stage, the mLYTl transcript amounts to 10.5 % and the transcript that gives origin to the kLYTl isoform represents 89.5 % of the mRNAs derived from the LYT1 gene [15]. Accordingly, in epimastigotes, it was observed that the kLYTl isoform was more abundant than the mLYTl one [16].
In this work, was carried out a detailed characterization of the 5’ and 3’ UTRs of the LYT1 mRNAs, as a first step of a project aimed to the identification of RBPs implicated in the modulation of both their expression and alternative trans-splicing rates. In this way, was experimentally determined the sequence of the 5’ UTR from both isoforms in two T. cruzi strains belonging to the DTU I and its conservation was analyzed in ll additional UTRs from parasites belonging to different DTUs. In addition, for the first time, the 3’ UTRs of the LYT1 mRNAs were delimitated; interestingly, the existence of two types of transcripts, differing in length by ll6 nts and generated by alternative polyadenylation was uncovered. The analysis was completed by a thoroughly search of structural motifs present in the different UTRs.
Materials and methods
Parasites
Epimastigotes of Trypanosoma cruzi 058PUJ [18] and D.A [MHOM/CO/200l/D.A.] [19], two DTU I isolates as well as of Y [20], a DTU II strain, were grown on Liver Infusion Tryptose Medium [LIT] supplemented with l0 % of fetal bovine serum [Eurobio, Inc., Les Ulis, France] at 26 °C. Trypomastigotes of the 058PUJ isolate were obtained at described elsewhere [21]. In brief, Green Monkey renal fibroblast-like cells (Vero cells; ATCC CCL-81, Manassas, VA) were cultured in DMEM (Eurobio, Les Ulis, France) supplemented with 10 % FBS (Eurobio), 2 mM L-glutamine, 100 U/ml penicillin, 100 mg/ml streptomycin, and 0.01 M HEPES (Eurobio, Les Ulis, France). The cells were grown at 37 °C in a humid atmosphere at 5 % CO2. When Vero cells reached semiconfluence, they were incubated for 10 h with T. cruzi epimastigotes (ratio 1:10, cell:parasites). Trypomastigotes were collected from the culture supernatant of infected Vero cells at 168 h postinfection. Infection of fresh Vero cells was then conducted with the trypomastigotes using the same infection multiplicity as before. After two additional passages, the trypomastigotes were collected and extensively washed thrice with 1 x PBS (Eurobio Les Ulis, France); finally, parasites were suspended at 1 x 107 parasites per ml to proceed with DNA or RNA isolation (see next section).
Nucleic acids extraction, PCR and cloning
Fifteen ml of epimastigote culture in logarithmic phase (2 x 107 parasites/ml) were harvested to obtain genomic DNA. Total DNA was isolated using the phenol-chloroform-isoamilic alcohol method [22]. Total RNA was obtained from epimastigotes cultured at logarithmic phase and trypomastigotes grown at 1 x 107 cells/ml, using the TRIzol method [Invitrogen, California, USA] [23]. The first-strand cDNA synthesis was carried out from total RNA using an oligo-dT primer and the Transcriptor first strand cDNA synthesis kit [Roche, Inc., Mannheim, Germany].
The 5’ and 3’ UTRs, and the upstream and downstream regions of the LYT1 gene were amplified by PCR using different sets of specific primers synthetized by IDT, Inc. [Miami, USA], [Suppl. 1]. One μL, containing 10 to 12 ng of cDNA, for amplification of the UTR regions or 100 ng of genomic DNA for amplification of the intergenic regions, was used. The PCR reaction mixes were performed in a final volume of 20 μl, containing: 1 x reaction buffer [10 mM Tris-HCl pH 9.0, 50 mM KCl, 0.1 % Triton X-100], 1.5 mM MgCl2, dNTPs (0.4 mM each) mix, 0.5 aM of each primer, 1.5 M betain, 0.06 units per [A of expand high fidelity Taq polymerase enzyme [Roche, Inc., Mannheim, Germany]. The PCR reactions were carried out in a thermal cycler C1000 Bio-Rad, under the following amplification profile: 95 °C/5 min [initial denaturation], 39 cycles at 92 °C/30 s, annealing at 48 °C - 62 °C/30s [Suppl. 1] and extension for 1 min at 72 °C, with a final incubation at 72 °C for 10 min.
The amplified fragments were resolved in agarose gels, stained with HydraGreen™ Safe Dye and visualized under UV light. The RT-PCR or PCR products were excised from gels, purified using Wizard® SV Gel and PCR Clean-Up System [Promega, Inc., Madison, WI, USA] and cloned into the pGEM-T Easy Vector [Promega, Inc., Madison, WI, USA]. The cloned sequences were determined by the sequencing Service of Macrogen Inc., Korea and their authenticity confirmed by comparison with the upstream and downstream sequences surrounding the LYT1 ORF. Finally, the sequences determined in this work were submitted to the GenBank database [Suppl. 2].
In silico analysis of the LYT1 mRNA UTRs of T. cruzi
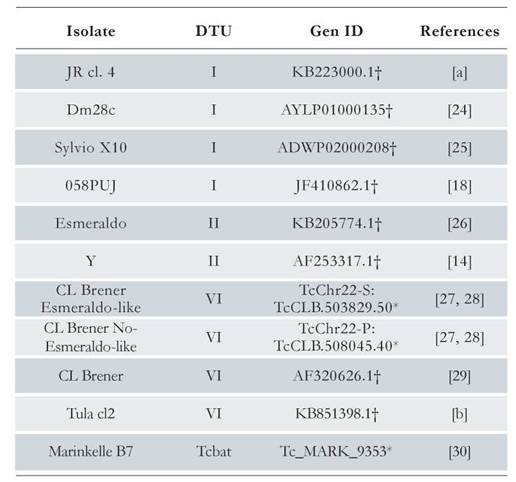
By using the genomic sequence of the LYT1 gene reported in the TritrypDB database [ID code TcCLB.503829.50] as query sequence, a BLAST search was performed in the TrytripDB and GenBank databases, obtaining eleven LYT1 sequences from different strains and DTUs (see Table 1), [24-30]. Through a multiple alignment using the Clustal Omega program [31], the LYT1 gene sequence was delimited in each of the sequences listed in Table 1.
With the purpose of searching RNA motifs in the twelve UTR regions, firstly, the secondary structure for conserved sequences located in the 5' and 3' UTRs was determined with the LocARNA program [32]. In addition, to identify potential functional RNA motifs, the RegRNA2.0 program [33] was used. Finally, in-silico predicted mRNA structures were generated for each of the twelve analyzed sequences by the RNAstructure program [34]. After gathering all this information, a consensus secondary structure of the functional motifs presents in all the isolates included in the study was generated using the RNAalifold server [35].
Results
Identification of the 5’ UTRs present in the specific mRNAs that code for the mLYT1 and kLYT1 isoforms
The mature mRNAs of the LYT1 gene in the T. cruzi CL Brener strain are generated in vivo by alternative trans-splicing, giving rise to three transcripts differing in their 5’ UTRs [15]. Two of them code for the mLYT1 isoform and the other for the kLYTl one. In order to determine whether or not an alternative trans-splicing for this gene also occurs in other T. cruzi strains, and to delimitate the 5’ UTRs (as the first step for identification of LYT1 mRNA interacting RBPs), these regions were mapped in two DTU I isolates, taking advantage of the presence of the common SL sequence at the 5’-end of the mRNAs. The results showed that as occurs in epimastigotes from the CL Brener strain, alternative trans-splicing also takes place in epimastigotes from the D.A. isolate and trypomastigotes from the 058PUJ isolate [Suppl. 2], demonstrating that the existence of two mRNAs species (kLYTl and mLYTl) derived from the LYT1 gene is a conserved feature among different strains of T. cruzi [Fig. 1a and 1b]. In contrast, it was not found any evidence on the third transcript, previously described in the CL Brener strain, that would correspond to a mRNA (coding for the mLYTl isoform), which is generated by using a non-canonical GG dinucleotide splicing acceptor site. As shown in Fig. 1a and 1b, the two types of 5’ UTRs are well conserved (both in sequence and length) when compared with the equivalent genomic regions in ten different T. cruzi strains (belonging to the DTU-I, II, VI, and Tcbat). A complete sequence alignment is provided as supplementary material, showing the existence of a high sequence conservation [Suppl. 3 and 4].
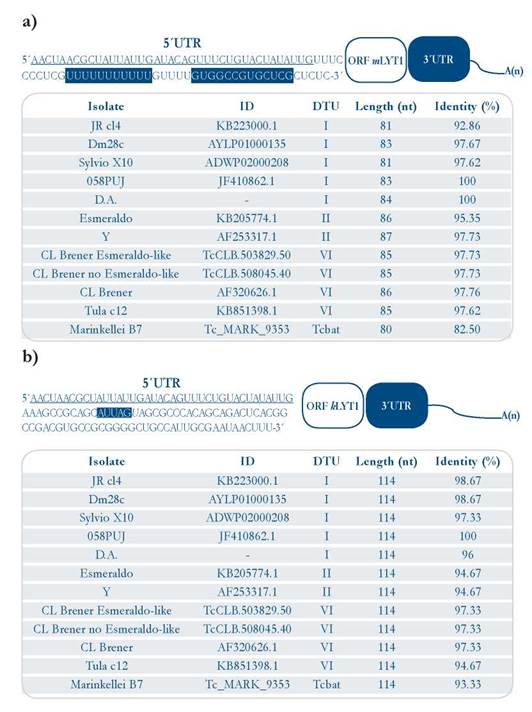
Figure 1 Sequences of the LYT1 5’ UTR mRNA isoforms. [a]. Upper panel: 5’ UTR otLYTI sequence from the 058PUJ (DTU I) isolate (GenBank KT279498). Lower panel: 5’ UTR otLYTI length sequences and sequence identity (using Clustal Omega) among twelve T. cruzi isolates [b]. Upper panel: 5’ UTR kLYTl sequence from the 058PUJ (DTU I) isolate (GenBank ID KT328589). Lower panel: 5’ UTR kLYTl length sequences and sequence identity among twelve T cruzi isolates The Leader Sequence Leader [SL] common to both 5’ UTRs is underlined, and the motifs present in each of the UTRs are highlighted in blue.
3’ UTR sequences of the LYT1 mRNA
With the aim of determining the 3’ UTR of the LYT1 mRNA, this region was initially amplified using RNA from epimastigotes and trypomastigotes of the 058PUJ isolate [Fig. 2]. Unexpectedly, three amplification products were clearly observed in both epimastigote and trypomastigote RNA samples. After cloning of the PCR products from the epimastigote sample, two different fragments were cloned, named here 3’ UTR-I and 3’ UTR-II, that after sequencing were found to be 264 and 380 nucleotides in length, respectively, without taking into account the poly (A) sequence [Fig. 3a and 3b]. Similar results were obtained when RNA from epimastigotes of the D.A isolate was used for RT-PCR amplification [suppl. 2]. However, after cloning of the amplification products obtained from the trypomastigote sample, only the fragment corresponding to the 3’ UTR-I region, was successfully cloned in the 058PUJ isolate [suppl. 2]. Nevertheless, according to the amplification bands observed in both parasite stages [Fig. 2], it might be concluded that both types of 3’-UTRs (and even a third one) are generated in equivalent proportions in epimastigotes and trypomastigotes.

Figure 2 Amplification from complementary DNA (cDNA) of the 3’ UTR of LYT1 of T. cruzi isolate 058PUJ by conventional PCR [a]. Upper panel: Schematic representation of the cloning strategy. [b]. Lower panel: cDNA from epimastigotes [lane E], Molecular Weight Marker 1 Kb plus Invitrogen™ [lane M], cDNA from trypomastigotes [lane T].

Figure 3 Sequences of two 3’ UTRs generated from transcription of the LYT1 gene in the 058PUJ (DTU I) isolate. [a]. Upper panel: 3’ UTR-I LYT1 sequence (deposited in GenBank under accession number KU973680), and sequence identity in twelve T. cruzi isolates [b]. Lower panel: 3’ UTR-II sequence (deposited in GenBank under accession number KU973678) and sequence identity percent in twelve T. cruzi isolates. The motifs present in each of the UTRs are highlighted in blue.
Comparison of the 3’ UTR-I with the LYT1 genomic corresponding sequence revealed that downstream at the end of the 3’ UTR-I, there exists in the genome a poly (A) stretch [suppl. 5]. Thus, in order to verify the existence of this 3’ UTR-I, a new forward primer, the LY3U167F oligonucleotide, priming at nucleotides 167 to 186 from both 3’ UTR [suppl. 1], and the ACAC-HindIII-Oligo [DT]25 primer were used in a more astringent RT-PCR reaction. In this way, a 127 bp fragment whose sequence aligned, as expected, with the 96 last nucleotides of the 3’ UTR-I sequence was amplified, corroborating the existence of the 3’ UTR-I. According to these results, the polyadenylation sites were mapped at positions 1.924 and 2.044 from LYT1 start codon of the CL Brener isolate [GenBank ID AF320626.1].
Similar to the 5’ UTRs, both 3’ UTR regions are well conserved among different parasite isolates [Fig. 3a and 3b]. Nonetheless, some micro heterogeneity, including transitions, transversions and insertions/deletions, were observed at some places within these regions [suppl. 6 - 9].
Identification of conserved RNA motifs in the LYT1 mRNA UTRs
Once, each of the LYT1 gene sequences listed in Table 1 were delimited, the hypothetical 5’ UTR of each isoform was located using the trans-splicing signals: the AG dinucleotide acceptor of the leader sequence [SL] and the polypyrimidine tract. Meanwhile, the delimitation of the LYT1 3’ UTR was based on the location of the TGA stop codon [suppl. 5]. In addition, to confirm these results, a sequence comparison was performed using the experimentally obtained UTR regions and equivalent genomic regions present in databases.
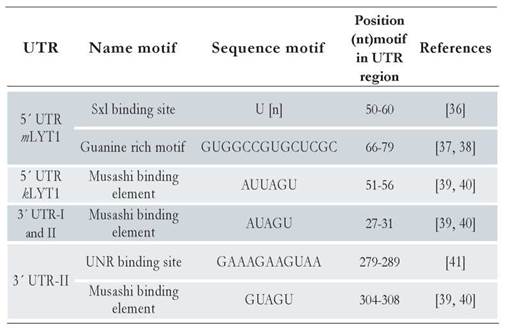
Following the pipeline described in Materials and Method section, several motifs were found in each UTR region of the different LYT1 mRNAs: two in the 5’ UTR mLYT1, one in the 5’ UTR kLYT1, one in the 3’ UTR-I and two in the 3’ UTR-II. Of note, the predicted motifs were different according to the UTR length. Indeed, a Sex-Lethal binding site (Sxl) and a Guanine rich region (G) were found in the 5’UTR mLYTl region. Meanwhile, in the 5’ kLYTl and 3’ UTR regions, a Musashi binding element, having a nucleotide variation between them, was detected. Besides this Musashi binding element common to both 3’ UTR regions, located within the last 116 nucleotides of the 3’UTR-II region, an Upstream of N-ras (UNR) element was predicted. All these motifs are commonly found in mRNAs from different eukaryotic organisms [36-41]. Table 2 lists the motifs identified and [suppl. 10] shows their structures and location within the predicted secondary structure of the two types of UTRs.
Discussion
Similar to alternative cis-splicing existing in most eukaryotes, alternative trans-splicing events may represent a source for protein diversity in trypanosomatids, including those with complex life cycles such as T. cruzi [42-44]. An example of this is the LYT1 gene, which codes for two protein isoforms, mLYT1 and kLYT1, whose expression is directed by an alternative trans-splicing occurring during the processing of the primary gene transcript [14,15]. This mechanism of gene expression gives rise to two protein isoforms that only differ in 28 out of the 552 aa comprising the larger isoform. Moreover, differences in the regulatory regions (i.e. UTRs) of the transcripts, through their interactions with a set of ribonucleoproteic complexes, are also linked to different cellular expression patterns, subcellular location and temporal function [45-47]. In this context, as the initial step within the project aimed to the identification of trans-acting regulatory proteins involved in the differential expression of LYT1 transcripts, in this work were precisely mapped the 5’ and 3’ UTRs of the LYT1 mRNAs. Thus, the existence of the LYT1 alternative trans-splicing event, previously reported for the CL Brener, a VI DTU isolate [15], was also confirmed in two DTU I T. cruzi isolates. This remarkable evolutionary conservation supports the importance of both isoforms for the parasite development and infectivity, independently of the parasite DTU [14,16]. Accordingly, the length and sequence of this region is well conserved among the different parasite DTUs analyzed. Moreover, in-silico analysis regarding the LYT1 presence in other organisms revealed that it is only present in the Trypanosoma genus (data not shown).
The UTRs, mainly the 3’ UTR regions, have been shown to play crucial roles in the posttranscriptional mechanisms of gene regulation [48]. Indeed, the knowledge and regulatory elements annotation of these regions could get insights into their role. In this work, was clearly demonstrated the existence in epimastigotes of two different 3’ UTRs in the LYT1 mRNAs that result from the use of different polyadenylation sites. In trypomastigotes, even though the fragment corresponding to the 3’ UTR-II could not be cloned, its existence is not ruled out by taking into account the RT-PCR results shown in Fig. 2. Alternative polyadenylation, which has been extensively described in all eukaryotic species, is a major mechanism of gene regulation, influencing both mRNA abundance and location [49,50]. Given the particular localization and differential expression of each LYT1 isoform, it is possible that each isoform derives from a transcript carrying a particular 3’ UTR. There are many examples showing the relevant role played by alternative 3’ UTRs as scaffolds to regulate protein location. For instance, the human CD47 protein is expressed from two mRNAs differing in the length of the 3’ UTR; the longest one facilitates its localization on the cell surface, and the shortest modulates its localization in the endoplasmic reticulum [50].
In trypanosomatids, gene expression is regulated at posttranscriptional level through mechanisms based on the interaction between RBPs and motifs, mostly present in the UTR regions [51,52]. Therefore, herein was searched for functional RNA motifs present in the different UTR regions identified in this work. As a result, several motifs in each type of UTR were found. Indeed, for the 5’ UTR mLYT1 two motifs were found. One of them, a guanine rich motif that has been described in other organisms as mediator a repressor effect on mRNA translation [38]. The other, a Sxl element, composed mainly of uracils, enhances the mRNA expression when present at the 5’ UTR of genes in tobacco plants and Drosophila [36,53]. Regarding the 5’ UTR kLYTl, a Musashi element was detected. These cis-elements regulate, positively or negatively, the translation of mRNAs through its recognition by specific RBPs that bind to the UAG core [54,55]. Interestingly, its presence was also observed in both types of LYTl 3’ UTRs. Meanwhile, the UNR motif, exclusive of the LYT1 3’UTR-II region, has been related to the destabilization of the mRNAs in mammals; this rich purine motif is bound by RBPs in RNP complexes that are engaged in rapid deadenylation of mRNAs [41].
Taking into account the different motifs identified in the LYT1 UTRs and the roles played in other organisms, it can be hypothesized the following regulatory model: The Sxl element would have a positive regulation on the mLYTl transcript in the trypomastigote stage whereas at the epimastigote stage this transcript would be down regulated by the G rich motif. In the same way, the Musashi elements would be positively regulating the mRNA expression of the kLYTl transcript at the epimastigote stage. Meanwhile, the UNR elements would be exerting a repressor effect on its translation at the trypomastigote stage. In addition, independently of the precise function of each of these motifs, surely they could have a pivotal role on the LYT1 isoforms expression.
Conclusion
The occurrence of LYTl trans-splicing in the parasite DTU I was confirmed, and predicted its existence in the parasite DTUs II, VI and Tcbat. For the first time, the 3’ UTR of the LYT1 mRNA was mapped showing two polyadenylation sites that give rise to two transcripts differing in 116 nts, which could have different roles on the location and expression patterns for each isoform. Moreover, different functional motifs associated with RNA metabolism have been identified in each one of the UTRs, and its potential involvement in the differential parasite-stage expression of each isoform has been discussed.
The supplements are available in http://revistas.javeriana.edu.co/index.php/scientarium/article/view/21288

