











 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
Introducción
Meuwissen, Hayes y Goddard (2001) propusieron el concepto de selección genómica que está basado en la distribución homogénea de miles de marcadores a lo largo del genoma y la estimación de sus efectos sobre características cuantitativas. Los avances tecnológicos y la disponibilidad de chips de polimorfismos de nucleótido simple (SNP, por sus siglas en ingles) de alta densidad han facilitado la implementación de modelos de evaluación genómica (Misztal, Legarra, & Aguilar, 2009). Estos modelos estiman efectos genéticos para los segmentos de cromosomas en una población que cuenta con información fenotípica y genómica, permitiendo conocer los efectos de cada SNP y estimar valores genómicos directos (Pintus et al., 2012).
Paralelamente, el crecimiento acelerado de datos genómicos y los cambios en programas de selección demandan una actualización constante de los sistemas de evaluación genética (VanRaden, 2016). Los modelos de selección genómica han incluido al mejor predictor lineal insesgado (BLUP, por sus siglas en ingles) ampliado, métodos Bayesianos y no paramétricos (Garrick, Dekkers & Fernando, 2014; Howard, Carriquiry & Beavis, 2014). La mayoría de estos modelos explican un mayor porcentaje de la varianza genética aditiva, incrementan las exactitudes de las estimaciones y reducen el intervalo generacional, contribuyendo notoriamente a un mayor progreso genético (Schaeffer, 2006). Por estas razones, la mayoría de estas aproximaciones generalmente han mostrado una superioridad respecto a los métodos de evaluación basados en el uso de la matriz de parentesco por pedigrí y que no incluyen información genómica (Legarra & Ducrocq, 2012).
El uso de estas herramientas genómicas esta relacionado con una mayor precisión en las estimaciones y consecuentemente un incremento en la ganancia genética. Las primeras evaluaciones genómicas contemplaron una metodología en múltiples etapas (msGBLUP). Sin embargo, valores genéticos no podían ser estimados para animales sin información genómica en msGBLUP (VanRaden, 2008). Por eso, Misztal et al. (2009) desarrollaron la metodología del mejor predictor lineal insesgado en una etapa (ssGBLUP) que incorpora información genómica y estima valores genéticos tanto para animales genotipados como para aquellos no genotipados. Por lo tanto, los objetivos de esta revisión fueron los siguientes: 1) describir la metodología ssGBLUP y sus avances; 2) identificar ventajas y limitaciones de la metodología, y 3) conocer la factibilidad de su uso en programas de mejoramiento genético con poblaciones genotipadas pequeñas.
Método
La consulta de la información bibliográfica se llevó a cabo en las bases de datos de Science Direct, Google Scholar, Scopus, NCBI y Scielo, a partir de 2001, año en que fue publicado el primer artículo de selección genómica referente al uso de SNP en evaluaciones genéticas. Las palabras usadas para la búsqueda fueron genomic evaluation, single step, genetic evaluation, genomic selection, genomic prediction, single nucleotide polymorphism y genetic modeling. La revisión no incluyo como fuentes bibliográficas libros ni artículos científicos publicados en un idioma diferente al inglés; tampoco fueron considerados artículos publicados en revistas no especializadas en genética y mejoramiento animal.
Desarrollo del tema
Los programas de mejoramiento genético convencionales han estado basados en la estimación de valores genéticos a partir de información genealógica y fenotípica (Chen et al., 2011). La metodología para estas evaluaciones ha sido el uso de modelos lineales mixtos, los cuales relacionan el desempeño productivo con efectos genéticos y son ajustados por efectos fijos y aleatorios no genéticos (Christensen, Madsen, Nielsen, Ostersen, & Su, 2012). Adicionalmente, la inclusión de la matriz de parentesco basada en el pedigrí permite estimar y ajustar valores genéticos de animales que carecen de información fenotípica, pero que están emparentados con la población evaluada. Sin embargo, el ajuste está basado en probabilidades promedio de segregación mendeliana y podría conducir en algunos casos a la estimación de valores genéticos idénticos para hermanos completos. Esto ignora el coeficiente de parentesco real y conduce a la subestimación o sobreestimación de valores genéticos para algunos animales.
El esquema anterior ha sido exitoso, pero el progreso genético obtenido es lento si la característica puede ser solamente medida en un sexo, después del sacrificio o tarde en la vida del animal. Para esto, esquemas de selección genómica permitieron incrementar considerablemente la tasa de ganancia genética (Meuwissen et al., 2001). Inicialmente, las evaluaciones genéticas con información genómica se realizaron a partir de paneles con miles de SNP, en el que todos los loci están asociados a características cuantitativas y se encuentran en desequilibrio de ligamiento con al menos un SNP (Goddard & Hayes, 2009). Por lo tanto, las decisiones de selección son basadas en los efectos de SNP de cada animal (Moser, Khatkar, Hayes, & Raadsma, 2010). En la actualidad, la selección genómica es ampliamente usada en ganado de carne y leche en países desarrollados como los Estados Unidos. Esto ha sido liderado por asociaciones de criadores y compañías privadas de inseminación artificial, cuyo objetivo es conducir hacia evaluaciones genéticas que mejoren la habilidad para identificar animales genéticamente superiores (Elzo et al., 2015).
Las primeras evaluaciones genómicas en el mundo usaron msGBLUP (figura 1), que consistió en lo siguiente: 1) la estimación de valores genéticos de manera convencional a través de un modelo animal, 2) estimación de los efectos de los SNP a partir de un grupo de animales con fenotipos y genotipos (población de referencia), y 3) la combinación de la información disponible bajo la teoría del índice de selección para obtener los valores genéticos genómicos (VanRaden, 2008). Sin embargo, la metodología de msGBLUP solo estima valores genéticos para animales genotipados y genera una alta variación de las confiabilidades (Garrick, Taylor, & Fernando, 2009). Aunque esta metodología permitió incrementar la precisión de las estimaciones para la selección de animales jóvenes (Konig & Swalve, 2009), esta predicción genómica tampoco considero el efecto de preselección, subestimando valores genéticos para animales jóvenes (Patry & Ducrocq, 2011).
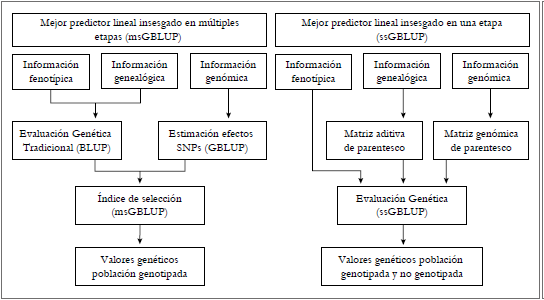
Adaptada de Aguilar et al. (2010) y Vanraden (2008)
Figura 1. Estructura de trabajo del mejor predictor lineal insesgado en múltiples etapas (msGBLUP) y en una sola etapa (ssGBLUP) para la estimación de valores genéticos.
Para superar dichos obstáculos, ssGBLUP fue desarrollada como una estructura de trabajo unificada que permite realizar estimaciones genómicas en toda la población y mas precisas que las obtenidas en msGBLUP (Aguilar et al., 2010) (figura 1).Adicionalmente, la rápida disminución en los costos de genotipificación permite aumentar el número de animales genotipados y robustecer mas los análisis a través de parentescos observados y no esperados como ocurre en la matriz de parentesco por pedigrí (Legarra, Aguilar, & Misztal, 2009). No obstante, esa tendencia podría no ser clara para países en vías de desarrollo, debido a la escasez de información genealógica, fenotípica y limitaciones económicas para el genotipado de una población grande.
Mejor predictor lineal insesgado en una etapa (ssGBLUP)
El ssGBLUP permite, a partir de la covarianza de frecuencias génicas, modelar el genotipo de un individuo a partir de la matriz de parentesco (Legarra & Ducrocq, 2012). Esta información es incluida en el sistema clásico de ecuaciones del modelo mixto, a través de la adición de una matriz de parentesco modificada y basada en las diferencias de parentesco genómico y por pedigrí de animales con genotipos (Fragomeni et al., 2015). El sistema de ecuaciones del modelo mixto que incluye la información genómica en ssGBLUP propuesto por Misztal et al. (2009) es el siguiente:
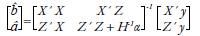
Donde b es un vector de soluciones para efectos fijos, â es un vector de soluciones para efectos aleatorios, y es un vector de observaciones, X es una matriz de incidencia que relaciona las observaciones con los efectos fijos, Z es una matriz de incidencia que relaciona las observaciones con el vector de efectos aleatorios genéticos aditivos directos, α es el cociente entre la varianza ambiental y la varianza genética aditiva directa y H es una matriz que combina parentescos por pedigrí y genómicos en una población en la que solamente algunos individuos están genotipados. Este modelo animal tiene la misma estructura propuesta por Henderson (1984), a excepción de la matriz de parentesco por pedigrí (A), la cual es reemplazada por la matriz H.
Igualmente, este modelo asume que los efectos de los marcadores tienen una distribución normal con varianza común (Legarra et al., 2009). La matriz H se obtiene a partir de la siguiente operación matricial:
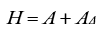
Donde A es la matriz de parentesco por pedigrí y AΔ contiene las diferencias de parentesco genómicas y aditivas de los animales genotipados:
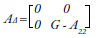
La estimación de la matriz genómica (G) es generada como RR´⁄ k, en donde R es una matriz que se obtiene a partir de (P - M), siendo M una matriz que contiene la información de cada marcador que los individuos heredaron (VanRaden, 2008), P es una matriz que contiene las frecuencias alélicas expresadas como una diferencia desde 0,5 y multiplicado por 2, siendo cada columna de P representada por 2( pi - 0,5); k es un escalar calculado a partir de 2Σ pi (1-pi), interpretando pi como la frecuencia del alelo de referencia para todos los casos. La sustracción G-A22 se realiza para evitar redundancia en la información de parentesco y la inflación en las estimaciones de los valores genómicos (Liu, Goddard, Reinhardt, & Reents, 2014; Misztal et al., 2013).
Aguilar et al. (2010) realizaron la primera evaluación genética con ssGBLUP en ganado Holstein en los Estados Unidos, obteniendo mayores precisiones. A partir de ese momento, ssGBLUP se ha convertido en un método más simple y más preciso para la estimación de valores genéticos. Sin embargo, para su implementación se debe genotipar una parte de la población, considerar exigencias computacionales y evaluar factores que podrían influir en las precisiones de las estimaciones como la cantidad de animales genotipados y su relación con la población evaluada.
Población genotipada en ssGBLUP
En general, el tamaño de una población genotipada en programas de selección genómica debe ser mayor cuando la población candidata a selección tiene una menor relación genética con la población genotipada (Meuwissen, 2009). Asimismo, la información genómica de los ancestros es importante para aumentar las precisiones (Mulder, Calus, Druet, & Schrooten, 2012), pero en ocasiones esto es difícil debido a que la información genómica generalmente proviene de los animales más jóvenes en la población. Específicamente para ssGBLUP, la construcción de la matriz G debería ser construida con base en las frecuencias alélicas de los animales fundadores de la población evaluada (Christensen et al., 2012). Sin embargo, la construcción de esta matriz no es siempre factible en la practica debido a que usualmente los animales fundadores no están genotipados.
Lourenco et al. (2015) sugirieron que la estrategia de genotipado en ssGBLUP debe ser dirigida hacia los animales con mayor importancia, que generalmente son los más antiguos y con mayor información genealógica y fenotípica. Consecuentemente, la composición y el tamaño de la población genotipada son factores que afectan la estructura de varianzas y covarianzas de los valores genéticos y con ello la precisión. Aunque en ocasiones las poblaciones genotipadas para ssGBLUP son pequeñas, es importante tener en cuenta que poblaciones genotipadas de mayor tamaño contribuirán mas para la estimación de efectos alélicos y con una mayor precisión (VanRaden, 2016). Actualmente, un mayor número de animales genotipados con una mayor cantidad de SNP es más accesible por la disminución de los costos de la genotipifación y una mayor eficiencia en métodos estadísticos y computacionales en procesos de imputación (Uemoto, Sasaki, Sugimoto, & Watanabe, 2015).
Aspectos computacionales
Los métodos basados en el BLUP genómico requieren la inversa de la matriz de parentesco genómica (Pocrnic, Lourenco, Mazuda, Legarra, & Misztal, 2016). El costo de la inversión para G y A de los animales genotipados es una función cubica del número de animales genotipados (Legarra & Ducrocq, 2012), condición que dificulta la convergencia de las evaluaciones. Experiencias con bases de datos con mas de un millón de animales incluidos mostraron que la convergencia de la evaluación genética depende de la matriz G usada, la cual es generalmente singular y no puede ser invertida sin procedimientos adicionales (Aguilar et al., 2010). Por esta razón, VanRaden (2008) definió G=RR´⁄ k con el objetivo de obtener la matriz G en una forma semidefinida positiva. A pesar de eso, la matriz aun puede ser singular cuando existen dos individuos con el mismo genotipo (gemelos idénticos), condición que causa singularidad en la matriz A. En el mismo estudio, se desarrolló un método basado en la ponderación de las matrices G y A de los animales genotipados, así:
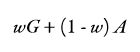
Donde w es el ponderador dado a la matriz genómica para facilitar las operaciones matriciales. Lo anterior evita problemas de singularidad y facilita la construcción de la matriz que contiene las diferencias entre parentesco por pedigrí y genómico de animales genotipados. Aunque w podría afectar las estimaciones, Aguilar et al. (2010) mostraron que valores de ponderación entre 0,95 y 0,98 no reportaron diferencias considerables y si genero una mayor precisión de la metodología ssGBLUP frente a msGBLUP.
La genotipificación masiva de animales permitió tener un mayor número de animales genotipados a la cantidad de SNP usada en el genotipado. Esta condición impidió de nuevo la inversión convencional de la matriz genómica, debido a que en ese escenario las matrices siempre son singulares (Liu, Goddard, Hayes, Reinhardt, & Reents, 2016). A partir de esto, varias aproximaciones computacionales han sido propuestas en los últimos anos, pero todas tienen problemas de convergencia o son de alto costo y complejas para su programación, en especial cuando se usan modelos multicaracter y regresiones aleatorias (Fragomeni et al., 2015). Inicialmente, la programación de ssGBLUP incluyo las ecuaciones previamente mostradas, pero esta aplicación estaría limitada para un número aproximado de hasta cien mil animales genotipados. Una cantidad superior dificulto hallar la inversa de la matriz genómica de parentesco debido a las limitaciones en memoria y tiempo de computación (Misztal, Legarra, & Aguilar, 2014; Misztal et al., 2013).
Ese obstáculo era predecible debido a que ssGBLUP permite incluir en su metodología todos los animales en la evaluación, exigiendo algoritmos más eficientes (Liu et al., 2014). Uno de los últimos avances para ssGBLUP propuestos por Misztal et al. (2014) fue el desarrollo del algoritmo para animales probados y jóvenes (APY), que divide la población genotipada en animales con registros y animales jóvenes. Este algoritmo hace uso de la recursión genómica para aproximar la inversa de la matriz genómica.
Las recursiones genómicas se pueden estimar a partir de la adición del coeficiente de parentesco genómico de cada animal y la varianza condicional (Misztal et al., 2014). Estas estimaciones están basadas en el supuesto de que existe un limitado número de segmentos de cromosomas o efectos de SNP que explican el total de la varianza genética (Pocrnic et al., 2016). El uso de este algoritmo redujo drásticamente los costos cuando el número de animales genotipados fue mayor a cincuenta mil y ayudo a la estabilidad del proceso iterativo. Incluso, la evaluación con un millón de animales genotipados es posible (Misztal et al., 2014).
Misztal (2016) mostro que, al usar la teoría de las recursiones genómicas apoyado en el algoritmo APY, la correlación entre las estimaciones halladas con el algoritmo frente a las halladas con la inversión directa y completa de la matriz genómica fueron superiores a 0,99. Por lo tanto, el algoritmo APY facilita la evaluación genómica, estimación de parámetros genéticos y estudios de asociación genómica en grandes poblaciones genotipadas. Por ejemplo, con una población mayor a quinientos mil animales, diez mil animales fueron suficientes en la recursión genómica para obtener estimaciones incluso más precisas que la inversión completa de la matriz genómica (Pocrnic et al., 2016).
Alternativas adicionales han incluido la implementación de un método indirecto de ssGBLUP apoyado en el algoritmo APY. Allí, los efectos de los SNP pueden ser calculados con el método convencional ssGBLUP, obteniendo un valor genómico directo. Después, este usa ponderaciones idénticas en toda la población para las diferentes fuentes de información, lo que disminuye el tiempo computacional requerido para estimar valores genéticos de animales jóvenes genotipados (Lourenco et al., 2014b). Así, una población genotipada mayor a cien mil animales podría presentar redundancia en los datos genómicos, por lo que un subconjunto de esta población y el uso de métodos indirectos podrían explicar la misma varianza genética estimada con toda la población. Aunque algunos problemas asociados a la dimensión de datos genómicos han sido solucionados, sin duda la selección genómica continuara evolucionando con los nuevos desarrollos en secuenciación y genotipificación (Garrick et al., 2014). No obstante, los algoritmos y recursos computacionales se están volviendo más limitantes que otros aspectos debido a que el número de animales y marcadores genotipados aumentan rápidamente (VanRaden, 2016).
Ventajas de ssGBLUP
El ajuste de las (co)varianzas entre individuos realizado en ssGBLUP permite obtener una derivación automática de las ponderaciones para la combinación de diferentes fuentes de información en la estimación de los valores genéticos, siendo una forma de evaluación genética mas practica y precisa, incluso para análisis multicaracter (Aguilar et al., 2010; Lourenco et al., 2014a). Además, las estimaciones a partir de modelos unicaracter o multicaracter con ssGBLUP permiten obtener valores genéticos para animales no genotipados, lo que no es posible en análisis con msGBLUP (Tsuruta, Misztal, Aguilar, & Lawlor, 2011).
Posiblemente, la precisión en las evaluaciones genéticas es el aspecto de mayor importancia, porque permite la clasificación adecuada de los animales y un mayor progreso genético si se asume la misma intensidad de selección. En selección genómica, un mecanismo por el cual aumentan las precisiones es a través de la validación y control de calidad del pedigrí (Christensen et al., 2012). ssGBLUP permite identificar conflictos en el pedigrí y ajustar relaciones de parentesco entre animales genotipados y no genotipados que conforman los archivos genealógicos de la población evaluada, disminuyendo sesgos en el ajuste de valores genéticos para animales sin información fenotípica.
Asimismo, los valores de consanguinidad son más precisos en ssGBLUP, porque la segregación mendeliana es estimada con eventos observados (SNP) y no con probabilidades medias esperadas incluidas en la matriz de parentesco por pedigrí (De Roos, Schrooten, Veerkamp, & Arendonk, 2011). Por ejemplo, dos animales genotipados que no tienen relación en la matriz de parentesco por pedigrí podrían mostrar relación en la matriz genómica debido a un ancestro en común que no fue registrado en el pedigrí. Igualmente, es posible que aparezcan mayores coeficientes de consanguinidad en los descendientes de animales genotipados tanto como estos estén relacionados (Legarra et al., 2009). La selección genómica puede tener entonces un rol fundamental en el control de la consanguinidad de las poblaciones, lo que permite un mayor control sobre la preservación de la diversidad genética de las poblaciones (Wensch-Dorendorf, Yin, Swalve, & Konig, 2011), y estimar con mayor precisión el efecto negativo de la consanguinidad sobre el desempeño fenotípico.
Por otro lado, una mayor cantidad de información conduce generalmente a una mayor exactitud de las estimaciones en programas de selección. Por esta razón, las precisiones y la varianza genética aditiva también aumentan cuando se usan chips de mas alta densidad ( Jattawa, Elzo, Koonawootrittriron, & Suwanasopee, 2015). Sin embargo, algunas poblaciones como la reportada por Loberg et al. (2015) explican un mayor porcentaje de la varianza genética cuando se utiliza la matriz de parentesco por pedigrí. Igualmente, Haile, Nieuwhof, Beard, Konstatinov y Hayes (2013) encontraron que la proporción genética de la varianza explicada por la información genómica y el pedigrí vario considerablemente entre características, sugiriendo que las discrepancias podrían ser dependientes de factores como la heredabilidad, e información fenotípica y genómica.
A pesar de que los chips de alta densidad no capturan toda la varianza genética, la precisión en la selección de animales jóvenes es mayor. Sin embargo, esta variación también depende de otros factores como el tamaño y la relación de parentesco de la población genotipada y no genotipada. Aunque el pedigrí en algunos casos explica una mayor proporción de la varianza, el error fue más alto en las estimaciones basadas en el pedigrí (Haile et al., 2013). Esto indica que, además de la población genotipada, los parámetros genéticos y la magnitud de sus estimaciones también podrían afectar las estimaciones genómicas, que están basadas en la (co)varianza encontrada entre los individuos de la población genotipada y su relación con animales no genotipados.
Respecto al componente económico, la reducción del intervalo generacional, el aumento en la precisión de los valores genéticos y una posible disminución en la toma de registros fenotípicos en empresas ganaderas han sido razones suficientes para justificar la implementación de programas de selección genómica. Schaeffer (2006) indico que la selección genómica podría incluso permitir la existencia de hatos en los que la toma de registros no sea necesaria, por lo que las decisiones de selección estarían hechas con base en información genómica. Por ahora, ssGBLUP parece ser la alternativa más eficiente para ajustar simultáneamente información de animales genotipados y no genotipados, en especial para países con un número bajo de animales genotipados, en los que el acceso a la genotipifacion de toda la población puede ser limitado e incluso nulo. Esto ubica a ssGBLUP como la metodología con más oportunidades e impactos económicos para reemplazar los sistemas clásicos de evaluación genética (Christensen et al., 2012).
Limitaciones de ssGBLUP
Los métodos estadísticos basados en la distribución de los efectos de los SNP pueden ser clasificados en dos grupos: el primero asume que los SNP tienen algún efecto sobre la característica y que la varianza de cada efecto es igual; el segundo permite a los marcadores tener diferentes tipos de distribución (Zhang et al., 2010). Las predicciones genómicas lineales se rigen bajo el modelo infinitesimal, que asume que todos los SNP contribuyen uniformemente con la varianza genética, por lo que no es posible identificar loci de gran efecto asociados a características cuantitativas (Goddard, 2009). La metodología ssGBLUP no tiene en cuenta los efectos mayores de loci dado el supuesto de efecto infinitesimal con el que fue construida ssGBLUP. Aunque el supuesto de varianza común permanece en continua discusión y parecería ser una desventaja, el trabajo con otro tipo de distribución y que contemplen loci de efecto mayor no ha presentado ganancias importantes en la precisión respecto a ssGBLUP.
Legarra y Ducrocq (2012) describieron que las tres limitantes más importantes que tiene ssGBLUP son las siguientes: 1) el costo de computación, 2) la necesidad de modificar el software existente, que cubre una variedad de modelos usados de evaluación, y 3) su forma lineal, que excluye los estimadores no lineales incluyendo información genómica. Aunque los desarrollos científicos y computacionales indicados en esta revisión han cubierto las dos primeras problemáticas, la discusión sigue presente en los supuestos que asume el modelo. Estudios con simulación han mostrado que las aproximaciones bayesianas han resultado en estimaciones de mayor precisión; sin embargo, evaluaciones con datos reales en producción de leche han terminado en algunas ocasiones con estimaciones de menor precisión (Su et al., 2012). Además, Aguilar et al. (2010) mostraron que al usar un numero grande de SNP con igual varianza fue apropiado para la mayoría de las características productivas.
A pesar de que el escenario más realista resulta cuando el modelo asume que existen genes de efecto mayor, estudios en los que se compararon las estimaciones genómicas a través de metodologías bayesianas y ssGBLUP mostraron que las precisiones y las pérdidas de varianza son muy similares (Wang, Misztal, Aguilar, Legarra, & Muir, 2012). Por lo tanto, ssGLUP tendría una mayor ventaja frente a otras metodologías que no permiten la inclusión de animales no genotipados y requieren datos fenotípicos para la derregresion de los valores genéticos. Esto limita la aplicación practica de la selección genómica en poblaciones en las que no es posible genotipar toda la población, condición que a menudo se presenta. Sin embargo, cuando el número de registros es bajo y la densidad de los chips usados es alta, otras metodologías parecen ser superiores a ssGBLUP (Lourenco et al., 2014b).
Uso de ssGBLUP en poblaciones pequeñas
El interés de la selección genómica es identificar un conjunto de SNP para incrementar la precisión de los valores genéticos. Para esto, es necesario estimar los efectos de los SNP a partir de la población genotipada (Misztal, 2016; Misztal & Legarra, 2016). Una de las alternativas más útiles que permite económicamente incrementar el número de animales genotipados han sido los genotipos imputados desde chips de varias densidades y que son combinados en una sola evaluación genómica para reducir los costos y aumentar la precisión. Por ejemplo, el número de animales genotipados en los Estados Unidos de la raza Holstein incremento considerablemente a más de 950.000 animales en 2016 (Misztal, 2016).
La precisión de la imputación y las estimaciones genómicas también se ven afectadas cuando el número de animales genotipados, el desequilibrio de ligamiento (< 0,2) y las relaciones genéticas entre las poblaciones genotipadas son bajas ( Jattawa et al., 2015; Vanraden et al., 2013), debido a que no se pueden estimar con precisión los segmentos de haplotipos compartidos entre los animales relacionados y consecuentemente predecir los genotipos de los otros SNP de animales genotipados con chips de más baja densidad.
Un aumento del número de marcadores para los animales genotipados también podría ser una estrategia; no obstante, esto tuvo efectos contradictorios en las precisiones. Al aumentar el número de SNP, la tasa de recombinación disminuye, razón por la cual la precisión aumenta. Sin embargo, un aumento en el número de marcadores sin un aumento en el número de fenotipos es contraproducente, debido a que la colinealidad podría confundir los efectos y disminuir la precisión (Muir, 2007). Esta alternativa tendría incluso mayores contradicciones en países con poblaciones limitadas, ya que generalmente la toma de registros fenotípicos también es escasa.
La construcción de estrategias para mejorar la predicción en poblaciones pequeñas debe surgir a partir de los conceptos teóricos de ssGBLUP para incrementar la confiabilidad. Estos requerimientos son los siguientes: 1) mas genotipos debe resultar en igual o mayor confiabilidad; 2) un animal joven que ha sido genotipado no debe crear información adicional para otros animales; 3) la información extra de que un animal joven con ancestros no genotipados pueda contribuir a la población genotipada debe ser pequeña o nula, y 4) valores adicionales de confiabilidad no pueden ser obtenidos a partir de animales de líneas o razas diferentes (Misztal et al., 2013).
Los países con poblaciones pequeñas y con ausencia de evaluaciones genéticas generalmente soportan programas de selección en la continua importación de material genético de poblaciones mejoradas (Andonov et al., 2016). En selección genómica, esto es más importante cuando la correlación del desempeño fenotípico entre diferentes países es alta (Vanraden, 2016), indicando que las estimaciones genómicas para animales provenientes de poblaciones pequeñas pueden ser derivados de los efectos de los SNP estimados en otras poblaciones genotipadas (Andonov et al., 2016), y el ajuste de la matriz de parentesco podría ser mayor dadas las contribuciones genéticas de toros usados en programas de inseminación artificial en países con poblaciones pequeñas.
El intercambio de información genómica para incrementar el tamaño de la población genotipada es una aproximación eficiente para incrementar la precisión de las estimaciones cuando el numero de animales genotipados es bajo (Wiggans et al., 2015). No obstante, cuando la población contiene animales locales e importados, registros fenotípicos son aun necesarios para aumentar la precisión de la progenie (Lourenco et al., 2014a). Un camino para fortalecer esta alternativa es la inclusión de hembras con alta contribución genética y sus desempeños fenotípicos (Mc Hugh, Meuwissen, Cromie, & Sonesson, 2011). La inclusión de hembras genotipadas es deseable porque son parte importante de los programas de mejoramiento al estar sometidas igualmente a un proceso de selección y aportar valores fenotípicos a través de su propio desempeño y su progenie. Además, permiten incrementar el tamaño de la población genotipada, convirtiéndolas en una alternativa para reducir los sesgos y aumentar la precisión (Tsuruta, Misztal, & Lawlor, 2013). Asimismo, la inclusión de hembras podría ajustar con mayor precisión los grados de parentesco a partir de los efectos de los marcadores (Loberg et al., 2015). A pesar de que Tsuruta et al. (2013) evaluaron la inclusión de hembras en una población genotipada que no tenía limitaciones en tamaño, un criterio de selección fue la inclusión de vacas con registros y valores genéticos similares a los toros incluidos. Por lo tanto, la inclusión de vacas en poblaciones pequeñas y con escasez de evaluaciones genéticas podría mejorar considerablemente la implementación de un programa de selección genómica a través de la metodología ssGBLUP.
Conclusiones
La metodología ssGBLUP es una aproximación practica y más accesible para incluir información genómica a través de una mayor exactitud de la (co) varianza genética entre individuos en programas de selección. Aunque las limitaciones más importantes de ssGBLUP parecen estar dirigidas hacia aspectos computacionales, los continuos desarrollos en programación han permitido el desarrollo e implementación de algoritmos para las estimaciones genómicas. Finalmente, es necesario incluir información genómica de hembras locales y toros extranjeros que permitan aumentar la conectividad genética de la población genotipada y no genotipada para obtener mejores estimaciones de los valores genéticos.

