










Services on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkCiencia y Tecnología Agropecuaria
Print version ISSN 0122-8706On-line version ISSN 2500-5308
Cienc. Tecnol. Agropecuaria vol.21 no.1 Mosquera Jan./Apr. 2020 Epub Dec 30, 2019
https://doi.org/10.21930/rcta.vol21_num1_art:1548
Genética, reproducción y mejoramiento animal
Genetic evaluations in cattle using the single-step genomic best linear unbiased predictor

1Docente e investigador, Universidad de Ciencias Aplicadas y Ambientales (UDCA), Facultad de Ciencias Agropecuarias. Bogotá, Colombia.
2Director de Investigación y Desarrollo, Corporación Colombiana de Investigacion Agropecuaria (AGROSAVIA). Mosquera, Colombia.
3Docente e investigador, Universidad de Antioquia, Facultad de Ciencias Agrarias, Escuela de Producción Agropecuaria, Grupo GaMMA. Medellín, Colombia.
Conventional genetic evaluations have been framed on estimated breeding values from equation systems of mixed models that consider simultaneously random and fixed effects. Recently, the development in genome sequencing technologies has allowed obtaining genomic information to include in genetic evaluations in order to increase the accuracy and genetic progress, and decrease the generation interval. The single-step best linear unbiased predictor is a methodology developed in the last years and accepts including genomic information replacing the genomic relationship matrix by a matrix that combines relationship by pedigree, and the genomic relationship of a genotyped population, allowing the estimation of breeding values for non-genotyped animals. The aim of this review article was to describe the methodology and its recent progress, as well as to know some of the strategies that could be used when the number of genotyped animals is low.
Keywords animal husbandry; genetic improvement; genetic markers; genomics; phenotypes
Las evaluaciones genéticas convencionales han estado enmarcadas en la estimación de valores genéticos a partir de los sistemas de ecuaciones de modelos mixtos que consideran efectos aleatorios y fijos simultáneamente. En los últimos anos, el desarrollo de tecnologías de secuenciación del genoma ha permitido obtener información genómica que puede ser incluida en las evaluaciones genéticas para incrementar las confiabilidades y el progreso genético, así como para disminuir el intervalo generacional. El mejor predictor lineal insesgado en una etapa es una metodología que incluye información genómica, reemplazando la matriz de parentesco por una matriz que combina el parentesco por pedigrí y el parentesco genómico de una población genotipada, permitiendo la estimación de valores genéticos para animales no genotipados. El objetivo de este artículo de revisión fue la descripción de la metodología y sus recientes avances, así como conocer algunas de las estrategias que podrían ser llevadas a cabo cuando el número de animales genotipados es bajo.
Palabras clave fenotipos; ganadería; genómica; marcadores genéticos; mejoramiento genético
Introduction
Meuwissen, Hayes and Goddard (2001) proposed the concept of genomic selection that is based on the homogeneous distribution of thousands of markers throughout the genome and the estimation of its effects on quantitative characteristics. Technological advances and the availability of high-density single nucleotide polymorphisms (SNPs) have facilitated the implementation of genomic evaluation models (Misztal, Legarra, & Aguilar, 2009). These models estimate genetic effects for chromosome segments in a population that has phenotypic and genomic information, allowing to know the effects of each SNP and estimate direct genomic values (Pintus et al., 2012).
At the same time, the accelerated growth of genomic data and the changes in selection programs demand a constant update of the genetic evaluation systems (VanRaden, 2016). Among the genomic selection models, the extended best linear unbiased predictor (BLUP), and the Bayesian and non-parametric methods stand out (Garrick, Dekkers & Fernando, 2014; Howard, Carriquiry & Beavis, 2014). Most of these models explain a higher percentage of the additive genetic variance, increase the accuracy of the estimates, and reduce the generational interval, contributing significantly to higher genetic progress (Schaeffer, 2006). For these reasons, most of these approaches have generally shown superiority over evaluation methods based on the use of the relationship matrix by pedigree, not including genomic information (Legarra & Ducrocq, 2012).
The use of these genomic tools is related to higher precision in the estimates and, consequently, an increase in genetic gain. The first genomic evaluations contemplated a multi-step methodology (msGBLUP). However, breeding values could not be estimated for animals without genomic information in msGBLUP (VanRaden, 2008). Therefore, Misztal et al. (2009) developed the single step genomic best linear unbiased predictor (ssGBLUP) methodology that incorporates genomic information and estimates breeding values for both genotyped and non-genotyped animals. Accordingly, the aims of this review were the following: 1) to describe the ssGBLUP methodology and its progress; 2) to identify advantages and limitations of the methodology, and 3) to know the feasibility of its use in breeding programs with small genotyped populations.
Method
Relevant bibliographic information was consulted in the following databases: Science Direct, Google Scholar, Scopus, NCBI, and Scielo, from the year 2001, i.e., the year in which the first genomic selection article that refers to the use of SNP in genetic assessments was published. The words used for the search were: genomic evaluation, single step, genetic evaluation, genomic selection, genomic prediction, single nucleotide polymorphism, and genetic modeling. The review did not include books or scientific articles published in a language other than English as bibliographic sources, nor articles published in journals not specialized in genetics and animal breeding.
Topic development
Conventional genetic improvement programs have been based on the estimation of breeding values from genealogical and phenotypic information (Chen et al., 2011). The methodology for these evaluations has been the use of mixed linear models, which relate productive performance to genetic effects and are adjusted for non-genetic fixed and random effects (Christensen, Madsen, Nielsen, Ostersen, & Su, 2012). Additionally, the inclusion of the pedigree-based relationship matrix allows estimating and adjusting the breeding values of animals that lack phenotypic information but are related to the population evaluated. However, the adjustment is based on average probabilities of Mendelian segregation and could, in some cases, lead to the estimation of identical breeding values for complete siblings. This ignores the real relationship coefficient and leads to underestimation or overestimation of breeding values for some animals.
The previous scheme has been successful, but the genetic progress obtained is slow if the characteristic can only be measured in one sex, after sacrifice or late in the life of the animal. For this, genomic selection schemes allowed to significantly increase the genetic gain rate (Meuwissen et al., 2001). Initially, genetic evaluations with genomic information were made from panels with thousands of SNPs, in which all loci were associated with quantitative characteristics and were in linkage imbalance with at least one SNP (Goddard & Hayes, 2009). Therefore, selection decisions are based on the SNP effects of each animal (Moser, Khatkar, Hayes, & Raadsma, 2010). Currently, genomic selection is widely used in meat and milk cattle in developed countries such as the United States. This has been led by breeder associations and private artificial insemination companies, whose objective leads towards genetic evaluations that improve the ability to identify genetically superior animals (Elzo et al., 2015).
The first genomic evaluations in the world used msGBLUP (figure 1), which consisted of the following: 1) estimation of breeding values in a conventional way through an animal model, 2) estimation of the effects of SNPs from a group of animals with phenotypes and genotypes (reference population), and 3) the combination of the information available under the theory of the selection index to obtain genomic breeding values (VanRaden, 2008). However, the msGBLUP methodology only estimates breeding values for genotyped animals and generates a high variation of reliabilities (Garrick, Taylor, & Fernando, 2009). Although this methodology allowed increasing the precision of the estimates for the selection of young animals (Konig & Swalve, 2009), this genomic prediction also did not consider the pre-selection effect, underestimating breeding values for young animals (Patry & Ducrocq, 2011).
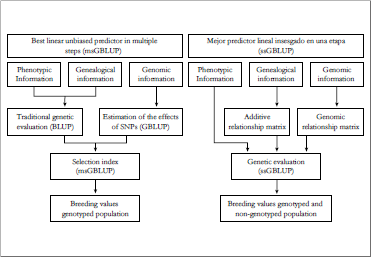
Adapted from Aguilar et al. (2010) and Vanraden (2008)
Figure 1. Working structure of the best linear unbiased predictor in multiple steps (msGBLUP) and in a single step (ssGBLUP) for the estimation of breeding values.
To overcome the obstacles mentioned above, ssGBLUP was developed as a unified work structure that allows genomic estimates to be made throughout the population and that are more accurate than those obtained in msGBLUP (Aguilar et al., 2010) (figure 1). Additionally, the rapid decrease in genotyping costs allows increasing the number of genotyped animals and further, strengthen the analyzes through observed and unexpected relationships as occurs in the relationship matrix by pedigree (Legarra, Aguilar, & Misztal, 2009). However, this trend may not be evident for developing countries due to the lack of genealogical, phenotypic and economic limitations for genotyping a large population.
Best linear unbiased predictor in one step (ssGBLUP)
ssGBLUP allows, from the covariance of gene frequencies, to model the genotype of an individual from the relationship matrix (Legarra & Ducrocq, 2012). This information is included in the classic system of equations of the mixed model, through the addition of a modified relationship matrix based on the differences of genomic relationship and pedigree of animals with genotypes (Fragomeni et al., 2015). The system of equations of the mixed model that includes the genomic information in ssGBLUP proposed by Misztal et al. (2009) is as follows:
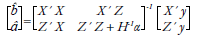
Where b is a vector of solutions for fixed effects, â is a vector of solutions for random effects, and y is a vector of observations; X is an incidence matrix that relates observations to fixed effects, Z is an incidence matrix that relates the observations with the vector of direct additive genetic random effects, α is the ratio between the environmental variance and the direct additive genetic variance, and H is a matrix that combines pedigree and genomic relationships in a population in which only some individuals are genotyped. This animal model has the same structure proposed by Henderson (1984), except for the relationship matrix by pedigree (A), which is replaced by matrix H.
Likewise, this model assumes that the effects of the markers have a normal distribution with a common variance (Legarra et al., 2009). Matrix H is obtained from the following matrix operation:
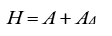
Where A is the relationship matrix by pedigree and AΔ contains the genomic and additive relationship differences of genotyped animals: 2( pi - 0,5)
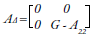
The estimation of the genomic matrix (G) is generated as RR´⁄ k, where R is a matrix that is obtained from (P - M), and P is a matrix that contains allelic frequencies expressed as a difference from 0.5 and multiplied by 2; each P column is represented by 2( pi - 0,5). Further, M is a matrix that contains the information of each marker that the individuals inherited (VanRaden, 2008); k is a scalar calculated from 2Σ pi (1-pi), interpreting pi as the frequency of the reference allele for all cases. The subtraction G-A22 is performed to avoid redundancy in relationship information and inflation in the genomic value estimates (Liu, Goddard, Reinhardt, & Reents, 2014; Misztal et al., 2013).
Aguilar et al. (2010) performed the first genetic evaluation with ssGBLUP in Holstein cattle in the United States, obtaining higher precision results. From that moment on, ssGBLUP has become a simpler and more accurate method for estimating breeding values. However, a part of the population must be genotyped for its implementation, considering computational requirements and evaluating factors that could influence the accuracy of the estimates, such as the number of genotyped animals and their relationship with the population evaluated.
Genotyped population in ssGBLUP
In general, the size of a genotyped population in genomic selection programs should be larger when the selected candidate population has a lower genetic relationship with the genotyped population (Meuwissen, 2009). Also, the genomic information of the ancestors is important to increase the accuracies (Mulder, Calus, Druet, & Schrooten, 2012), but sometimes this is difficult because the genomic information usually comes from the youngest animals in the population. Specifically, for ssGBLUP, the construction of the G matrix should be based on the allelic frequencies of the founding animals of the evaluated population (Christensen et al., 2012). However, the construction of this matrix is not always feasible in practice because normally, the founding animals are not genotyped.
Lourenco et al. (2015) suggested that the genotyping strategy in ssGBLUP should be directed towards the most important animals, which are generally the oldest and with most genealogical and phenotypic information. Consequently, the composition and size of the genotyped population are factors that affect the structure of variances and covariances of breeding values, and with it, the precision. Although genotyped populations for ssGBLUP are sometimes small, it is crucial to keep in mind that larger genotyped populations will contribute more to the estimation of allelic effects and with higher precision (VanRaden, 2016). Currently, a higher number of genotyped animals with a more significant number of SNPs is more accessible due to the decrease in genotyping costs and higher efficiency in statistical and computational methods in imputation processes (Uemoto, Sasaki, Sugimoto, & Watanabe, 2015).
Computational aspects
Methods based on genomic BLUP require the inverse of the genomic relationship matrix (Pocrnic, Lourenco, Mazuda, Legarra, & Misztal, 2016). The investment cost to construct G and A matrices of genotyped animals is a cubic function of the number of genotyped animals (Legarra & Ducrocq, 2012), a condition that makes the convergence of evaluations difficult. Experiences with databases with more than one million animals included showed that the convergence of the genetic evaluation depends on the G matrix used, which is generally unique and cannot be reversed without additional procedures (Aguilar et al., 2010). Therefore, VanRaden (2008) defined G=RR´⁄ k aiming at obtaining the G matrix in a semi-definite positive form. Despite this, the matrix can still be unique when there are two individuals with the same genotype (identical twins), a condition that causes singularity in matrix A. In the same study, a method based on the weighting of G and A matrices of genotyped animals was developed, as follows:
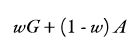
Where w is the weighting given to the genomic matrix to facilitate matrix operations. This avoids singularity problems and facilitates the construction of the matrix that contains the differences between pedigree and genomic relationship of genotyped animals. Although w could affect the estimates, Aguilar et al. (2010) showed that weighting values between 0.95 and 0.98 did not report significant differences and did generate higher accuracy of the ssGBLUP methodology versus msGBLUP.
Mass genotyping of animals allowed a higher number of animals genotyped, considering the amount of SNPs used in genotyping. This condition, again, prevented the conventional inversion of the genomic matrix, because, in that scenario, the matrices are always unique (Liu, Goddard, Hayes, Reinhardt, & Reents, 2016). From this point, several computational approaches have been proposed in recent years, but all have convergence problems or are expensive and complex to be programmed, especially when using multi-character models and random regressions (Fragomeni et al., 2015). Initially, ssGBLUP programming included the equations previously shown, but this application would be limited for an approximate number of up to one hundred thousand genotyped animals. A higher quantity made it difficult to find the inverse of the genomic relationship matrix due to limitations in memory and computation time (Misztal, Legarra, & Aguilar, 2014; Misztal et al., 2013).
This obstacle was predictable because ssGBLUP allows including in its methodology all the animals in the evaluation, demanding more efficient algorithms (Liu et al., 2014). One of the latest advances for ssGBLUP proposed by Misztal et al. (2014), was the development of the algorithm for tested and young animals (ATY), which divides the genotyped population into animals with records and young animals. This algorithm makes use of genomic recursion to approximate the inverse of the genomic matrix.
Genomic recurrences can be estimated from the addition of the genomic relationship coefficient of each animal and the conditional variance (Misztal et al., 2014). These estimates are based on the assumption that there is a limited number of chromosome segments or SNP effects that explain the total genetic variance (Pocrnic et al., 2016). The use of this algorithm drastically reduced costs when the number of genotyped animals was higher than fifty thousand and helped with the stability of the iterative process. Even an evaluation with one million genotyped animals is possible (Misztal et al., 2014).
Misztal (2016) showed that, when using the genomic recursion theory supported by the ATY algorithm, the correlation between the estimates found with the algorithm versus those found with the direct and complete inversion of the genomic matrix was higher than 0.99. Therefore, the ATY algorithm facilitates genomic evaluation, estimation of genetic parameters, and studies of genomic association in large genotyped populations. For example, with a population larger than five hundred thousand animals, ten thousand animals were sufficient in genomic recursion to obtain even more accurate estimates compared to the complete inversion of the genomic matrix (Pocrnic et al., 2016).
Additional alternatives have included the implementation of an indirect method of ssGBLUP supported by the ATY. In this, the effects of SNPs can be calculated using the conventional ssGBLUP method, obtaining a direct genomic value. Then, identical weights are used throughout the population for different sources of information, which decreases the computational time required to estimate the breeding values of genotyped young animals. (Lourenco et al., 2014b). Thus, a genotyped population larger than one hundred thousand animals could show redundancy in genomic data, so a subset of this population and the use of indirect methods could explain the same estimated genetic variance with the entire population. Although some problems associated with the genomic data dimension have been solved, there is no doubt that the genomic selection will continue to evolve with new developments in sequencing and genotyping (Garrick et al., 2014). However, algorithms and computational resources are becoming more limiting than other aspects because the number of animals and genotyped markers increase rapidly (VanRaden, 2016).
Advantages of ssGBLUP
The adjustment of the (co)variances between individuals carried out in ssGBLUP allows obtaining an automatic derivation of the weights for the combination of different sources of information in the estimation of the breeding values, being a more practical and precise form of genetic evaluation, even for multi-character analyses (Aguilar et al., 2010; Lourenco et al., 2014a). Besides, estimates based on single or multi-character models with ssGBLUP allow obtaining breeding values for non-genotyped animals, which is not possible in analyzes with msGBLUP (Tsuruta, Misztal, Aguilar, & Lawlor, 2011).
Possibly, the precision in genetic evaluations is the most important aspect, because it allows the proper classification of animals and greater genetic progress if the same selection intensity is assumed. In genomic selection, a mechanism by which precision is increased is through the validation and quality control of the pedigree (Christensen et al., 2012). ssGBLUP allows identifying conflicts in the pedigree and adjust relationship relationships between genotyped and non-genotyped animals that comprise the genealogical archives of the population evaluated, reducing biases in the adjustment of breeding values for animals without phenotypic information.
Likewise, consanguinity values are more precise in ssGBLUP, because Mendelian segregation is estimated with observed events (SNPs) and not with expected average probabilities included in the relationship matrix by pedigree (De Roos, Schrooten, Veerkamp, & Arendonk, 2011). For example, two genotyped animals that have no relationship in the relationship matrix by pedigree could show a relationship in the genomic matrix due to a common ancestor that was not recorded in the pedigree. Likewise, higher inbreeding coefficients may appear in the descendants of genotyped animals as much as they are related (Legarra et al., 2009). Genomic selection can then have a fundamental role in the control of the consanguinity of populations, which allows greater control over the conservation of the genetic diversity of populations (Wensch-Dorendorf, Yin, Swalve, & Konig, 2011), and estimate more accurately the negative effect of consanguinity on phenotypic performance.
On the other hand, a more considerable amount of information generally leads to higher accuracy of the estimates in selection programs. For this reason,the accuracies and additive genetic variance also increase when higher density chips are used (Jattawa, Elzo, Koonawootrittriron, & Suwanasopee, 2015). However, some populations, such as the one reported by Loberg et al. (2015) explain a higher percentage of the genetic variance when using the relationship matrix by pedigree. Similarly, Haile, Nieuwhof, Beard, Konstatinov and Hayes (2013) found that the genetic proportion of the variance explained by the genomic information and the pedigree varied considerably between characteristics, suggesting that the discrepancies could be dependent on factors such as heritability, as well as phenotypic and genomic information.
Although high-density chips do not capture the entire genetic variance, the accuracy in the selection of young animals is higher. However, this variation also depends on other factors such as the size and relationship of the genotyped and non-genotyped population. Although the pedigree, in some cases, explains a more significant proportion of the variance, the error was higher in the estimates based on the pedigree (Haile et al., 2013). This indicates that, in addition to the genotyped population, genetic parameters and the magnitude of their estimates could also affect genomic estimates, which are based on the (co)variance found between individuals in the genotyped population and their relationship with non-genotyped animals.
Regarding the economic component, the reduction of the generational interval, the increase in the accuracy of breeding values, and a possible decrease in the collection of phenotypic records in livestock companies have been enough reasons to justify the implementation of genomic selection programs. Schaeffer (2006) indicated that genomic selection could even allow the existence of herds in which registration is not necessary so that selection decisions would be made based on genomic information. For now, ssGBLUP seems to be the most efficient alternative to simultaneously adjust information of genotyped and non-genotyped animals, especially for countries with a low number of genotyped animals, in which access to genotyping of the entire population can be limited and even null. This places ssGBLUP as the methodology with the most opportunities and economic impacts to replacing the traditional genetic evaluation systems (Christensen et al., 2012).
Limitations of ssGBLUP
Statistical methods based on the distribution of the effects of SNPs can be classified into two groups: the first assumes that SNPs have some effect on the characteristic and that the variance of each effect is the same; the second allows markers to have different types of distribution (Zhang et al., 2010). Linear genomic predictions are governed by the infinitesimal model, which assumes that all SNPs contribute uniformly to the genetic variance, so it is not possible to identify large-effect loci associated with quantitative characteristics (Goddard, 2009). The ssGBLUP methodology does not consider the major effects of loci given the assumption of the infinitesimal effect with which ssGBLUP was constructed. Although the assumption of common variance remains in continuous discussion and would seem to be a disadvantage, working with another type of distribution and contemplating major effect loci has not presented significant gains in accuracy compared to ssGBLUP.
Legarra and Ducrocq (2012) described that the three most important limitations that ssGBLUP has are the following: 1) the computational cost, 2) the need to modify existing software that covers a variety of used evaluation models, and 3) its linear form, which excludes non-linear estimators including genomic information. Although scientific and computer developers mentioned in this review have covered the first two issues, the discussion is still present in the assumptions assumed by the model. Simulation studies have shown that Bayesian approaches have resulted in more accurate estimates; however, when evaluating real milk production data, lower accuracy estimates have sometimes been obtained (Su et al., 2012). In addition, Aguilar et al. (2010) showed that using a large number of SNPs with equal variance was appropriate for most of the productive characteristics.
Although the most realistic scenario is when the model assumes that there are genes of more significant effect, studies in which genomic estimates were compared through Bayesian methodologies and ssGBLUP, showed that the accuracies and losses of variance are very similar (Wang, Misztal, Aguilar, Legarra, & Muir, 2012). Therefore, ssGLUP would have a more significant advantage over other methodologies that do not allow the inclusion of non-genotyped animals and require phenotypic data for the breakdown of breeding values. This limits the practical application of genomic selection in populations in which it is not possible to genotype the entire population, a condition that often occurs. However, when the number of records is low, and the density of the chips used is high, other methodologies appear to be better compared to ssGBLUP (Lourenco et al., 2014b).
Use of ssGBLUP in small populations
The interest of genomic selection is to identify a set of SNPs to increase the accuracy of the breeding values. For this, it is necessary to estimate the effects of SNPs from the genotyped population (Misztal, 2016; Misztal & Legarra, 2016). One of the most useful alternatives that economically allow increasing the number of genotyped animals has been the genotypes imputed from chips of various densities and that are combined in a single genomic evaluation to reduce costs and increase accuracy. For example, the number of genotyped animals of the Holstein breed in the United States increased considerably to more than 950,000 animals in 2016 (Misztal, 2016).
The imputation accuracy and genomic estimates are also affected when the number of genotyped animals, the linkage imbalance (< 0.2), and the genetic relationships between genotyped populations are low ( Jattawa et al., 2015; Vanraden et al., 2013). This occurs because the haplotype segments shared among related animals cannot be accurately estimated, and consequently, predict the genotypes of the other SNPs of animals genotyped with lower density chips.
An increase in the number of markers for genotyped animals could also be a strategy; however, this had contradictory effects on the accuracies. As the number of SNPs increases, the recombination rate decreases, and this is the reason why the accuracy increases. However, an increase in the number of markers without an increase in the number of phenotypes is counterproductive because collinearity could confuse the effects and decrease accuracy (Muir, 2007). This alternative would have even greater contradictions in countries with limited populations since generally obtaining phenotypic records is also scarce.
The construction of strategies to improve prediction in small populations should arise from the theoretical concepts of ssGBLUP to increase reliability. These requirements are as follows: 1) more genotypes should result in equal or higher reliability; 2) a young animal that has been genotyped should not create additional information for other animals; 3) the extra information that a young animal with non-genotyped ancestors can contribute to the genotyped population must be small or null, and 4) additional reliability values cannot be obtained from animals of different lines or races (Misztal et al., 2013).
Countries with small populations and the absence of genetic evaluations generally support selection programs in the continuous import of genetic material from improved populations (Andonov et al., 2016). In genomic selection, this is more important when the correlation of phenotypic performance between different countries is high (Vanraden, 2016), indicating that genomic estimates for animals from small populations may be derived from the effects of SNPs estimated in other genotyped populations (Andonov et al., 2016). Furthermore, the adjustment of the relationship matrix could be greater given the genetic contributions of bulls used in artificial insemination programs in countries with small populations.
The exchange of genomic information to increase the size of the genotyped population is an efficient approach to increase the accuracy of estimates when the number of genotyped animals is low (Wiggans et al., 2015). However, when the population contains local and imported animals, phenotypic records are still necessary to increase the accuracy of the progeny (Lourenco et al., 2014a). One way to strengthen this alternative is the inclusion of females with high genetic contribution and their phenotypic performances (Mc Hugh, Meuwissen, Cromie, & Sonesson, 2011).
The inclusion of genotyped females is desirable because they are an important part of breeding programs as they are also subject to a selection process and provide phenotypic values through their own performance and progeny. In addition, they allow increasing the size of the genotyped population, making them an alternative to reduce biases and increase accuracy (Tsuruta, Misztal, & Lawlor, 2013). Also, the inclusion of females could more accurately adjust the degrees of relationship based on the effects of the markers (Loberg et al., 2015). Although Tsuruta et al. (2013) evaluated the inclusion of females in a genotyped population that had no limitations in size, a selection criterion was the inclusion of cows with records and breeding values similar to the included bulls. Therefore, the inclusion of cows in small populations and with a lack of genetic evaluations could significantly improve the implementation of a genomic selection program through the ssGBLUP methodology.
Conclusions
The ssGBLUP methodology is a practical and more accessible approach to include genomic information through higher accuracy of the genetic (co)variance between individuals in selection programs. Although the most important limitations of ssGBLUP seem to be directed towards computational aspects, the continuous developments in programming have allowed the development and implementation of algorithms for genomic estimations. Finally, it is necessary to include genomic information of local females and foreign bulls that allow increasing the genetic connectivity of the genotyped and non-genotyped population to obtain better estimates of breeding values.
Acknowledgments
To Asociacion Colombiana de Criadores de Ganado Simmental (Asosimmental) (Colombian Association of Simmental cattle breeders) and the Genetics Department of AGROSAVIA in Tibaitata within the framework of the cooperation agreement TV16-11 that allowed the implementation of the project “Information system based on the development of growing performance tests for the identification of high genetic merit reproducers in the Simmental and Simbrah races.” Further, to the sustainability call of Universidad de Antioquia obtained by the GaMMA research group. Finally, the first author acknowledges Universidad de Ciencias Aplicadas y Ambientales (UDCA) and Colciencias (call 727 of 2015) for the financial support for his training in the PhD in Animal Sciences of the Universidad de Antioquia.
REFERENCES
Aguilar, I., Misztal, I., Johnson D., Legarra, A., Tsuruta, S., & Lawlor, T. (2010). Hot topic: A unified approach to utilize phenotypic, full pedigree, and genomic information for genetic evaluation of Holstein final score. Journal of Dairy Science, 93(2), 743-752. doi:10.3168/jds.2009-2730. [ Links ]
Andonov, S., Lourenco, D. A. L., Fragomeni, B. O., Masuda, Y., Pocrnic, I., Tsuruta, S., & Misztal, I. (2016). Accuracy of breeding values in small genotyped populations using different sources of external information-a simulation study. Journal of Dairy Science, 100(1), 395-401. doi:10. 3168/jds.2016-11335. [ Links ]
Chen, J., Wang, Y., Zhang, Y., Sun, D., Zhang, S., & Zhang, Y. (2011). Evaluation of breeding programs combining genomic information in Chinese Holstein. Agricultural Sciences in China, 10(12), 1949-1957. doi:10.1016/ S1671-2927(11)60196-X. [ Links ]
Christensen, O., Madsen, P., Nielsen, B., Ostersen, T., & Su, G. (2012). Single-step methods for genomic evaluation in pigs. Animal, 6(10), 1565-1571. doi:10.1017/S175173 1112000742. [ Links ]
De Roos, A. P. W., Schrooten, C., Veerkamp, R. F., & Van Arendonk, J. A. M. (2011). Effects of genomic selection on genetic improvement, inbreeding, and merit of young versus proven bulls. Journal of Dairy Science, 94(3), 1559-1567. doi:10.3168/jds.2010-3354. [ Links ]
Elzo, M. A, Thomas, M. G, Johnson, D. D., Martinez, C. A., Lamb, G. C., Rae, D. O., & Driver, J. D. (2015). Genetic parameters and predictions for direct and maternal growth traits in a multibreed Angus-Brahman cattle population using genomic-polygenic and polygenic models. Livestock Science, 178, 43-51. doi:10.1016/j.livsci.2015.06.015. [ Links ]
Fragomeni, B. O., Lourenco, D. A. L., Tsuruta, S., Masuda, Y., Aguilar, I., Legarra, A., & Misztal, I. (2015). Hot topic: Use of genomic recursions in single-step genomic Best Linear Unbiased Predictor (BLUP) with a large number of genotypes. Journal of Dairy Science, 98(6), 4090-4094. doi:10.3168/jds.2014-9125. [ Links ]
Garrick, D., Dekkers, J., & Fernando, R. (2014). The evolution of methodologies for genomic prediction. Livestock Science, 166(1), 10-18. doi:10.1016/j.livsci.2014.05.031. [ Links ]
Garrick, D., Taylor, J. F., & Fernando, R. L. (2009). Deregressing estimated breeding values and weighting information for genomic regression analyses. Genetics Selection Evolution, 31, 41-55. doi:10.1186/1297-9686-41-55. [ Links ]
Goddard, M. (2009). Genomic selection: prediction of accuracy and maximisation of long-term response. Genetica, 136(2), 245-257. doi:10.1007/s10709-008-9308-0. [ Links ]
Goddard, M. E., & Hayes, B. J. (2009). Mapping genes for complex traits in domestic animals and their use in breeding programmes. Nature Reviews, 10(6), 381-391. doi: 10.1038/nrg2575. [ Links ]
Haile, M., Nieuwhof, G. J., Beard, K. T., Konstatinov, K. V., & Hayes, B. J. (2013). Comparison of heritabilities of dairy traits in Australian Holstein-Friesian cattle from genomic and pedigree data and implications for genomic evaluations. Journal of Animal Breeding and Genetics, 130(1), 20-31. doi:10.1111/j.1439-0388.2013.01001.x. [ Links ]
Henderson, C. R. (1984). Applications of linear models in animal breeding. Guelph, Canada: University of Guelph. [ Links ]
Howard, R., Carriquiry, A. L., & Beavis, W. D. (2014). Parametric and nonparametric statistical methods for genomic selection of traits with additive and epistatic genetic architectures. Genes Genomes Genetics, 4(6), 1027-1046. doi:10.1534/g3.114.010298. [ Links ]
Jattawa, D., Elzo, M. A., Koonawootrittriron, S., & Suwanasopee, T. (2015). Comparison of genetic evaluations for milk yield and fat yield using a polygenic model and three genomicpolygenic models with different sets of SNP genotypes in Thai multibreed dairy cattle. Livestock Science, 181, 58-64. doi:10.1016/j.livsci.2015.10.008. [ Links ]
Konig, S., & Swalve, H. H. (2009). Application of selection index calculations to determine selection strategies in genomic breeding programs. Journal of Dairy Science, 92(10), 5292-5303. doi:10.3168/jds.2009-2232. [ Links ]
Legarra, A., Aguilar, I., & Misztal, I. (2009). A relationship matrix including full pedigree and genomic information. Journal of Dairy Science, 92(9), 4656-4663. doi:10.3168/jds.2009-2061. [ Links ]
Legarra, A., & Ducrocq, V. (2012). Computational strategies for national integration of phenotypic, genomic, and pedigree data in a single-step best linear unbiased prediction. Journal of Dairy Science, 95(8), 4629-4645. doi:10.3168/jds.2011-4982. [ Links ]
Liu, Z., Goddard, M. E., Hayes, B. J., Reinhardt, F., & Reents, R. (2016). Technical note: Equivalent genomic models with a residual polygenic effect. Journal of Dairy Science, 99(3), 2016-2025. doi:10.3168/jds.2015-10394. [ Links ]
Liu, Z., Goddard, M. E., Reinhardt, F., & Reents, R. (2014). A single-step genomic model with direct estimation of marker effects. Journal of Dairy Science, 97(9), 5833-5850. doi:10.3168/jds.2014-7924. [ Links ]
Loberg, A., Durr, J. W., Fikse, W. F., Jorjani, H., & Crooks, L. (2015). Estimates of genetic variance and variance of predicted genetic merits using pedigree or genomic relationship matrices in six Brown Swiss cattle populations for different traits. Journal of Animal Breeding and Genetics, 132(5), 376-385. doi:10.1111/jbg.12142. [ Links ]
Lourenco, D. A. L., Misztal, I., Tsuruta, S., Aguilar, I., Ezra, E., Ron, M., & Weller, J. I. (2014a). Methods for genomic evaluation of a relatively small genotyped dairy population and effect of genotyped cow information in multiparity analyses. Journal of Dairy Science, 97(3), 1742-1752. doi:10.3168/jds.2013-6916. [ Links ]
Lourenco, D. A. L., Misztal, I., Tsuruta, S., Aguilar, I., Lawlor, T. J., Forni, S., & Weller, J. I. (2014b). Are evaluations on young genotyped animals benefiting from the past generations? Journal of Dairy Science, 97(6), 3930-3942. doi:10.3168/jds.2013-7769. [ Links ]
Lourenco, D. A. L., Tsuruta, S., Fragomeni, B. O., Masuda, Y., Aguilar, I., Legarra, A., & Misztal, I. (2015). Genetic evaluation using single-step genomic best linear unbiased predictor in American Angus. Journal of Dairy Science, 93(6), 2653-2662. doi:10.2527/jas.2014-8836. [ Links ]
Mc Hugh, N., Meuwissen, T. H. E., Cromie, C. R., & Sonesson, A. K. (2011). Use of female information in dairy cattle genomic breeding programs. Journal of Dairy Science, 94(8), 4109-4118. doi:10.3168/jds.2010-4016. [ Links ]
Meuwissen, T. H. E. (2009). Accuracy of breeding values of “unrelated” individuals predicted by dense SNP genotyping. Genetics Selection Evolution, 41, 35. doi:10.1186/1297-9686-41-35. [ Links ]
Meuwissen, T. H. E., Hayes, B. J., & Goddard, M. E. (2001). Prediction of total genetic value using genome-wide dense marker maps. Genetics, 157(4), 1819-1829. [ Links ]
Misztal, I. (2016). Inexpensive computation of the inverse of the genomic relationship matrix in populations with small effective population size. Genetics, 202(2), 401-409. doi:10.1534/genetics.115.182089. [ Links ]
Misztal, I., & Legarra, A. (2016). Invited review: Efficient computation strategies in genomic selection. Animal, 11(5), 731-736. doi:10.1017/S1751731116002366. [ Links ]
Misztal, I., Legarra, A., & Aguilar, I. (2009). Computing procedures for genetic evaluation including phenotypic, full pedigree, and genomic information. Journal of Dairy Science, 92(9), 4648-4655. doi:10.3168/jds.2009-2064. [ Links ]
Misztal, I., Legarra, A., & Aguilar, I. (2014). Using recursion to compute the inverse of the genomic relationship matrix. Journal of Dairy Science, 97(6), 3943-3952. doi:10.3168/jds.2013-7752. [ Links ]
Misztal, I., Tsuruta, S., Aguilar, I., Legarra, A., Vanraden, P. M., & Lawlor, T. J. (2013). Methods to approximate reliabilities in single-step genomic evaluation. Journal of Dairy Science, 96(1), 647-654. doi:10.3168/jds.2012-5656. [ Links ]
Moser, G., Khatkar, M. S., Hayes, B. J., & Raadsma, H. W. (2010). Accuracy of direct genomic values in Holstein bulls and cows using subsets of SNP markers. Genetics Selection Evolution, 42(1), 37. doi: 10.1186/1297-9686-42-37. [ Links ]
Muir, W. M. (2007). Comparison of genomic and traditional blup-estimated breeding value accuracy and selection response under alternative trait and genomic parameters. Journal of Animal Breeding and Genetics, 124(6), 342-355. doi:10.1111/j.1439-0388.2007.00700.x [ Links ]
Mulder, H. A., Calus, M. P. L., Druet, T., & Schrooten, C. (2012). Imputation of genotypes with low-density chips and its effect on reliability of direct genomic values in Dutch Holstein cattle. Journal of Dairy Science, 95(2), 876-889. doi:10.3168/jds.2011-4490. [ Links ]
Patry, C., & Ducrocq, V. (2011). Evidence of biases in genetic evaluations due to genomic preselection in dairy cattle. Journal of Dairy Science, 94(2), 1011-1020. doi:10.3168/jds.2010-3804. [ Links ]
Pintus, M., Gaspa, G., Nicolazzi, E., Vicario, D., Rossoni, A., Ajmone-Marsan, P., & Macciotta, N. P. (2012). Prediction of genomic breeding values for dairy traits in Italian Brown and Simmental bulls using a principal component approach. Journal of Dairy Science, 95(6), 3390-3400. doi:10.3168/jds.2011-4274. [ Links ]
Pocrnic, I., Lourenco, D. A. L., Masuda, Y., Legarra, A., & Misztal, I. (2016). The dimensionality of genomic information and its effect on genomic prediction. Genetics, 203(1), 573-581. doi:10.1534/genetics.116.187013. [ Links ]
Schaeffer, L. R. (2006). Strategy for applying genome-wide selection in dairy cattle. Journal of Animal Breeding and Genetics, 123(4), 218-223. doi:10.1111/j.1439-0388.2006.00595.x. [ Links ]
Su, G., Madsen, P., Nielsen, U. S., Mantysaari, E. A., Aamand, G. P., Christensen, O. F., & Lund, M. S. (2012). Genomic prediction for Nordic Red cattle using one-step and selection index blending. Journal of Dairy Science, 95(2), 909-917. doi:10.3168/jds.2011-4804. [ Links ]
Tsuruta, S., Misztal, I., Aguilar, I., & Lawlor, T. J. (2011). Multiple-trait genomic evaluation of linear type traits using genomic and phenotypic data in US Holsteins. Journal of Dairy Science, 94(8), 4198-4204. doi:10.3168/jds.2011-4256. [ Links ]
Tsuruta, S., Misztal, I., & Lawlor, T. J. (2013). Short communication: genomic evaluations of final score for US Holsteins benefit from the inclusion of genotypes on cows. Journal of Dairy Science, 96(5), 3332-3335. doi:10.3168/jds.2012-6272. [ Links ]
Uemoto, Y., Sasaki, S., Sugimoto, Y., & Watanabe, T. (2015). Accuracy of high-density genotype imputation in Japanese Black cattle. Animal Genetics, 46(4), 388-394. doi:10.1111/age.12314. [ Links ]
Vanraden, P. M. (2008). Efficient methods to compute genomic predictions. Journal of Dairy Science, 91(11), 4414-4423. doi:10.3168/jds.2007-0980. [ Links ]
Vanraden, P. M. (2016). Practical implications for genetic modeling in the genomics era. Journal of Dairy Science, 99(3), 2405-2412. doi:10.3168/jds.2015-10038. [ Links ]
Vanraden, P. M., Null, D. J., Sargolzaei, M., Wiggans, G. R., Tooker, M. E., Cole, J. B., & Doak, G. A. (2013). Genomic imputation and evaluation using high-density Holstein genotypes. Journal of Dairy Science, 96(1), 668-678. doi:10.3168/jds.2012-5702. [ Links ]
Wang, H., Misztal, I., Aguilar, I., Legarra, A., & Muir, W. M. (2012). Genome-wide association mapping including phenotypes from relatives without genotypes. Genetics Research, 94(2), 73-83. doi:10.1017/S0016672312000274. [ Links ]
Wensch-Dorendorf, M., Yin, T., Swalve, H. H., & Konig, S. (2011). Optimal strategies for the use of genomic selection in dairy cattle breeding programs. Journal of Dairy Science, 94(8), 4140-4151. doi:10.3168/jds.2010-4101. [ Links ]
Wiggans, G. R., Su, G., Cooper, T. A., & Nielsen, U. S., Aamand, G. P., Guldbrandtsen, B., & Vanraden, P. M. (2015). Short communication: Improving accuracy of Jersey genomic evaluations in the United States and Denmark by sharing reference population bulls. Journal of Dairy Science, 98(5), 3508-3513. doi:10.3168/jds.2014-8874. [ Links ]
Zhang, Z., Liu, J., Ding, X., Bijma, P., de Koning, D. J., & Zhang, Q. (2010). Best linear unbiased prediction of genomic breeding values using a trait-specific marker-derived relationship matrix. PlosONE, 5(9), 1-8. doi:10.1371/journal.pone.0012648. [ Links ]
Received: December 27, 2018; Accepted: August 13, 2019
 Esta obra está bajo una Licencia Creative Commons Atribución-NoComercial-CompartirIgual 4.0 Internacional.
Esta obra está bajo una Licencia Creative Commons Atribución-NoComercial-CompartirIgual 4.0 Internacional.
