











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCTION
The Bovine Leukemia Virus (BLV) is a retrovirus of the Retroviridae family, Orthoretrovirinae subfamily, and Deltaretrovirus genus. It is known to be a type C retrovirus with global distribution; it induces persistent lymphocytosis and in a number of cases, it produces lymphoid tumors in cattle 1. It is an infectious, chronic and insidious disease that shows a low percentage of animals with clinical manifestations. The virus requires a long incubation period, generally from 3 to 5 years, and only a small proportion of animals develop tumors (0.1-5%) 2.
Like other complex retroviruses, the BLV genome is flanked by two identical long terminal repeats (LTRs) and contains structural genes gag, pol and env coding respectively, for the viral capsid protein, the RNA-dependent DNA polymerase (the reverse transcriptase) and the envelope protein 3. The env gene encodes for transmembrane gp51 and the envelope glycoprotein gp30, which are essential for viral infectivity and for inducing an immune response in naturally infected cattle 4,5.
The infectivity and/or pathogenicity of BLV may be affected by alterations in the env gene, which is considered as one of the most variable regions of the BLV genome 5. The classification of BLV strains into several different genotypes depends on the several restriction sites that this gene contains 6. In addition, it has conserved regions that are involved in the interaction between target cells and the virus. Thus, the differences in virus infectivity, BLV dissemination capacity and progression of illness may be attributed to minor changes in amino acid or nucleotide composition of the envelope protein 7,8.
The env gene codifies for the surface envelope glycoprotein gp51 of BLV, which is subjected to immune pressures and selection processes. For this reason, this gene has been found to be appropriate for phylogenetic analysis 9.
The analysis of BLV gp51 env gene sequences from different geographic regions has revealed the presence of different genetic groups that correlated with the geographic origin of the isolates 5. Currently, seven genotypes have been identified in South America based on the phylogenetic analysis of the env gene (10.
The aim of this study was to analyze BLV isolates by partial sequencing of the env gene in BLV isolates from ELISA positive dairy cattle from Pasto, Nariño, Colombia.
MATERIALS AND METHODS
Bovine samples
Blood samples were collected on FTA(tm) WHATMAN(r) cards from 18 BLV positive cattle (determined by indirect-ELISA), 24 months old or more, and their 13 daughters from three dairy farms in Pasto (southwestern region of Colombia). Blood was extracted by venipuncture of the coccygeal vein, and a small drop was placed in each FTA card. The cards were conserved in a cool and dark place until processed.
DNA extraction
DNA was obtained according to the Whatman(r) protocol: First, a sample disc (3mm) from the dried spot was cut and placed in a PCR amplification tube, then washed with 500μL of sterile water by pulse vortexing 3 times for 5 seconds (the tube should be given moderate manual mixing to disrupt the debris and to aid in washing). The disc was transferred to a new tube containing 30μL of sterile saline solution, ensuring that the disc was completely submerged, and heated at 95 °C for 15 to 30 min. The tube was pulse vortexed approximately 60 times. After that, it was centrifuged for 30 seconds to separate the matrix from the eluate that contains the purified DNA (Whatman FTA Elute Cards. Indicating FTA Elute procedure). The supernatant was refrigerated until processed.
Env amplification
In order to confirm the seropositive cattle, a fragment of the env gene was amplified by nested PCR in a Verity DNA Thermal Cycler (Invitrogen). PCR reactions were carried out according to the Virology lab protocol at Universidad Nacional del Centro (8). The first amplification round was briefly developed with 1 of template DNA obtained from the FTA cards, and 24 of master mix 1. Inner and outer primer sequences were as described in the literature 11. An initial incubation step at 94 °C for 5 min was performed, followed by 24 cycles, each consisting of denaturation at 95 °C for 30 s, annealing at 57°C for 30 s and extension step at 72 °C for 30 s (external primers, forward and reverse respectively, env5032 5'-TCTGTGCCAAGTCTCCCAGATA-3', env-5608 5'-AACAACAACCTCTGGGAAGGGT-3'). An aliquot of 3 of the amplified product was directly transferred to a new tube with fresh PCR mix (22 μl) for the second amplification round, under the same conditions, but annealing temperature was raised to 68 °C (internal primers forward and reverse respectively env5099 5'-GCGAGGCCGGGTCCAGAGCTGG-3', env5521 5'-AACAACAACCTCTGGGAAGGGT-3'), followed by a final extension at 72 °C for 10 min. Each master mix consisted in 1x reaction buffer, 1 μg/μl BSA, 1.5 mM MgCl2, 0.4 mM dNTP, 25 pM of each primer and 0.2 Units of Taq polymerase.
Amplified products were visualized on 0.9% agarose gels in TBE 0.5X buffer by staining with ethidium bromide, and sequencing of the amplified env gene fragment was performed. Briefly, amplification products were purified directly from the reaction tube using a PureLinkTM PCR Purification Kit (Invitrogen Cat K3100-01) according to the manufacturer's instructions. All of the amplified products were sequenced using the ABI PRISM Dye Terminator Cycle Sequencing Ready Reaction Kit and an ABI377 Genetic Analyzer (Applied Biosystems).
Data Analysis
We obtained thirty complete sequences ofthe env fragment, which were compared with full genome sequences available in the GenBank database. We were able to determine the genotypes present in the analyzed farms. The analysiswas carried out by the neighbor-joining method, using the Molecular Evolutionary Genetics Analysis (MEGA) software package version 4.0. The sequences were aligned using the ClustalW program and the Phylogenetic tree was built with an application of the same program.
Percentages of similarity of aligned nucleotide fragments of env gene were calculated. We could define clusters: one that comprises most of the sequences we have obtained, regardless of the group they belonged to or the location they came from, and another one that includes relationship between sampled animals.
RESULTS
Specificity of the serological determination: Of the 31 Indirect-ELISA positive animals, 27 sera were confirmed by nested PCR. We had enough material to be sequenced from only 17/27 amplified fragments. The homology among the obtained sequences and compared to the already published sequences was greater than 96%, demonstrating the specificity of the amplification reaction. In Figure 1, we can observe the amino acid alignment of the predicted sequence of the amplified region, compared to the reference sequence GenBank accession number K02120 12. Substitution of asparagine (N) by aspartic acid (D) is observed in nine of the sequences isolated in Pasto, Nariño, related to the Japanese sequence.

This unique change in the amino acidic sequence allowed us to build a new phylogenetic tree with only two branches, compared to the reference strain (K02120) (Figure 2).

The optimal tree with the sum of branch length = 0.03099057 is shown (Figure 2). The tree is drawn to scale, with branch lengths (next to the branches) in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the Maximum Composite Likelihood method and are in the units of the number of base substitutions per site. The analysis involved 17 nucleotide sequences. Codon positions included were 1st+2nd+3rd+Noncoding. All positions containing gaps and missing data were eliminated. There was a total of 331 positions in the final dataset.
A phylogenetic tree was built by comparing the nucleotide sequences of the proviruses fragments amplified in this work with the same region of published full-length BLV genomes. The sequence of the amplified fragments did not differ greatly from those already described, but allowed us to locate them in two clusters, BLV genotype 1, together with the strains from Australia, Costa Rica, Japan, Argentina, USA and Brazil, and BLV genotype 2, homologous to an Argentinian strain (Figure 3).
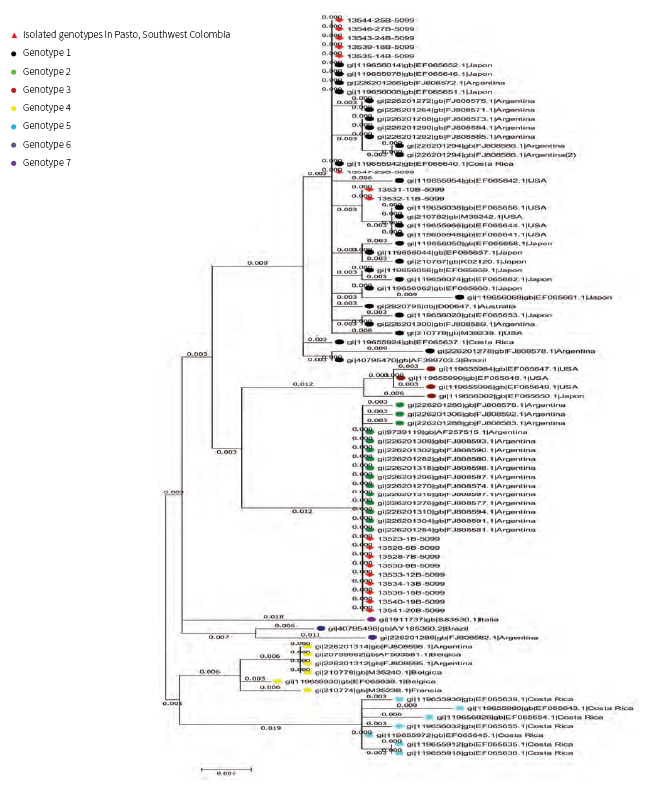
Figure 3 Phylogenetic tree based on partial nucleotide sequences of the BLV env gene from the different strains isolated for this work, compared to the same region of the other published full-genome sequences
In farm A we identified two positive animals (adult cows) with genotype 1. On the other hand, in farm B we found two dams and two daughters of each genotype (1 and 2). Finally, in farm C we found one dam and one daughter, which carry genotype 1, and two dams and three daughters, which carry genotype 2.
DISCUSSION
In this study, we analyzed the presence and genotypes of bovine leukemia virus throughout dairy farms of Pasto. A nested-PCR technique and direct sequencing of the env gene PCR product were developed to detect the presence of BLV Two genotypes were identified-genotype 1 and genotype 2. We can observe that genotype 1 is widely distributed around the world, but genotype 2 is more common in South American countries like Argentina and Colombia. According to the results obtained in other countries: (a) Iran reports only genotype 1 13; (b) in 2010, genotype 1 was identified as the most prevalent genotype in Japan 14; (c) in Jordan, genotype 1 and genotype 6 were identified. Matsumara et al. identified two genotypes of BLV in Turkey: genotype 1 and an unassigned genotype (15; (d) in South America, all seven BLV genotypes have been detected: genotype 1 in Uruguay; in Argentina, three different genotypes co-circulate (1,2,4) (9; (e) at least four different genotypes (1,5, 6) and (f) 16 in Brazil, although genotype 1 was still the most prevalent 15.
The env sequences of the BLV provirus from different locations worldwide have been classified previously into two and four genetic groupings. Some of these groups appear to correlate with the geographical origin of the strains 5,13,16,17. Rodríguez et al. believe that spreading of BLV genotypes could be due to the fact that the diversification of the virus has been driven by the historical dispersion of its host, which is associated with human migration patterns 9.
Furthermore, the occurrence of long-distance dispersal achieved by a small fraction of the virus population also supports the concept of viral transmission via close contact among individuals (9. The presence of more than one single genotype of BLV in different geographic areas may be explained by modern cattle trading tendencies5,7. In the area of study, we can associate the presence of two genotypes with reproductive management practices because, traditionally, improvement genetic programs use imported semen from the United States, Europe and Canada.
Only one genotype was detected in Farm A and two genotypes were found in two other farms (farms B and C). Farm A is a closed herd (it produces own replacements) and biosafety management is very carefully followed in veterinary practices. Therefore, the origin of BLV infection was one infected cow and the BLV genome of the gp51 region was rather stable and has been maintained among the cattle population of the farm.
Moreover, one cow died in farm A, and the necropsy showed multiple lesions like neoplasia in several visceral organs. In this farm, we only found genotype 1, previously associated with tumor samples in Japan (14). However, when seven genotypes were described, they deduced there was no apparent relation between the genotype of the infecting viral strains and the clinical manifestations9.
Farms B and C acquire animals from others dairy farms. Thus, the BLV infection in these farms most probably originated from several infected cows and could not be assigned mutations of BLV. The survey by Licursi et al. showed that no plural genotype was detected in individual animals when there was more than one genotype in the same farm 7.
On the other hand, some BLV variants are associated with the failure of antibody detection by standard serological methods and it has been proposed that sequence variability of BLV strains may result in antigenic differences that could affect viral serological identification in infected cattle populations from different geographic areas 11. In fact, it has been shown that the sensitivity and specificity of different serological testing methods already show a significant variation 18. However, other studies reported that there is no correlation between the BLV genotype and the serological status of infected animals and propose that the difference in the antiviral immune response may be related to the infection stage and other host factors (7.
We used Indirect ELISA to identify positive animals if the BLV variability affects the ability to identify viruses in all infected populations; the drawback of ELISA test is giving false negative results, mainly because of the weak humoral immune response sometimes mounted to the infection, but also because of a lack of sensitivity of this technique. As described in the literature, it is possible that the ELISA gives false positives, where n-PCR determination is highly specific and produce different results when compared with molecular methods 19. On the other hand, it was reported that there is a genetic variant of BLV and infected cattle remained seronegative by AGID and ELISA in all the cases 11. In addition, BLV-infected cattle are sometimes transiently seronegative under certain circumstances, such as early infections, per parturient state and co-infection with other viruses 20.
Finally, we conclude that distinct BLV provirus genotypes are found in different geographical areas and epidemiological analysis based on the variations of the gp51 region 21. The combination of sequencing and phylogenetic tree analysis was useful for characterization of BLV, because serologic diversity among BLV has not been identified 22.
Our results demonstrated that two BLV provirus genotypes (1 and 2) are present in the cattle of Pasto herds, and they show that different BLV provirus genotypes can be found in different geographical areas. This is one of the first attempts to assess the epidemiological situation of BLV in Colombia, using a molecular approach. We can suggest that this situation could be the result of independent introduction events that occurred from imported cattle from the USA and Europe many years ago. Knowledge of BLV genotypes circulating in Southern Colombia will be important for implementing appropriate cattle management policies and for developing appropriate anti-viral strategies and vaccines suitable for the region.

