










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Biotecnología
Print version ISSN 0123-3475
Rev. colomb. biotecnol vol.15 no.1 Bogotá Jan./June 2013
ARTÍCULO DE INVESTIGACIÓN
Expresión y purificación de las proteínas estructurales del rotavirus VP5* y VP8* en bacterias E. coli BL21(DE3)
Expression and purification of rotavirus structural proteins VP5* and VP8* in bacteria E. coli BL21(DE3)
Luz Yurany Moreno1 , Carlos Arturo Guerrero2 y Orlando Acosta3.
1Lic., MSc. en Bioquímica. Laboratorio Biología Molecular de Virus, Facultad de Medicina, Universidad Nacional de Colombia. E-mail: lymorenor@unal.edu.co. Carrera 30 No. 45-03, Facultad de Medicina, Bogotá, Colombia. Tel.: (57-1) 3165000 Ext. 15052 - 15053.
2MD., MSc., PhD en Cienicas Biomédicas. Profesor Titular, Departamento de Ciencias Fisiológicas, Facultad de Medicina-Instituto de Biotecnología, Universidad Nacional de Colombia, Bogotá, DC., Colombia. Autor de correspondencia: Carlos A. Guerrero, e-mail: caguerrerof@unal.edu.co, Carrera 30 No. 45-03, Facultad de Medicina, Bogotá, Colombia. Tel.: (57-1) 3165000 Ext. 15053, Fax: (57-1) 3165000 Ext. 15047.
3Bsc., MSc., PhD in Molecular Virology. Profesor Asociado, Departamento de Ciencias Fisiológicas, Facultad de Medicina-Instituto de Biotecnología, Universidad Nacional de Colombia, Bogotá, DC., Colombia. E-mail: oacostal@unal.edu.co. Carrera 30 No. 45-03, Facultad de Medicina, Bogotá, Colombia. Tel.: (57-1) 3165000 Ext. 15053, Fax: (57-1) 3165000 Ext. 15047.
Recibido: febrero 15 de 2013 Aprobado: junio 22 de 2013
Resumen
La caracterización de las proteínas estructurales del rotavirus y de las proteínas de la superficie de la célula hospedera implicadas en la unión y penetración del virion requiere de la disponibilidad de cantidades suficientes y de alto grado de pureza de estas proteínas. Por lo tanto, el objetivo de este trabajo fue expresar y purificar las proteínas estructurales del rotavirus de la cepa RRV, VP5* y VP8*, y producir anticuerpos policlonales dirigidos contra ellas. Se expresaron las proteínas recombinantes VP5* (rVP5*) y VP8* (rVP8*) en bacterias E. coli BL21(DE3) transfectadas con el plásmido pGEX-4T que contenía sus secuencias codificantes. Se consideraron como variables el medio de crecimiento, número de bacterias antes de inducir la expresión, concentración del inductor y tiempo de inducción. La mayor proporción de rVP8* se obtuvo cuando las bacterias transformadas se cultivaron en medio LB y la inducción se llevó a cabo con 1 mM de IPTG cuando el cultivo alcanzó una OD 600 nm de 0.5 y la inducción se mantuvo durante 6 h. rVP5* alcanzó la mayor proporción cuando células a una OD 600 nm de 0.2 fueron inducidas con 0.5 mM de IPTG durante 4 h en medio 2XYT en presencia de glucosa al 2 %. Las proteínas recombinantes obtenidas, acumuladas en la fracción insoluble, fueron solubilizadas con detergentes iónicos y no iónicos, seguido de purificación mediante cromatografía de afinidad antes de ser empleadas como antígenos para la producción de anticuerpos policlonales en conejos. Estos anticuerpos se caracterizaron mediante su capacidad de reconocimiento de los antígenos correspondientes en ELISA, "Western blotting", y en ensayos de inmunocitoquímica en células infectadas con rotavirus RRV. La cantidad y el grado de pureza de las proteínas recombinantes obtenidas, y los anticuerpos dirigidos contra ellas, anticipan su utilidad como herramientas en la caracterización de la interacción virus-célula.
Palabras clave: rotavirus, VP5*, VP8*, proteínas recombinantes, E. coli BL21(DE3)
Abstract
The characterization of rotavirus structural proteins and the cell surface proteins involved in virion binding and penetration depends on the availability of substantial amounts of highly purified proteins. The aim of the present work was to express and purify the rotavirus structural proteins VP5* and VP8* in order to produce polyclonal antibodies against them. Recombinant proteins VP5* (rVP5*) and VP8* (rVP8*) were expressed in E. coli cells transfected with pGEX-4T or pET 28a containing their corresponding encoding sequences. Culture medium, bacterial concentration before induction, inductor concentration and induction time were used as variables. The greater proportion of rVP8* was obtained when transfected bacteria were grown in medium LB and induction was started by adding 1 mM IPTG to cells at OD 600 nm 0.5 followed by 6 h-induction. The highest proportion of rVP5* was reached when cells at OD 600 nm 0.2 were induced with 0.5 mM IPTG for 4 h in medium 2XYT containing 2% glucose. The recombinant proteins accumulated in the insoluble fraction were solubilized with ionic and non-ionic detergents, and purified by affinity chromatography before being used as antigens for production of rabbit polyclonal antibodies. The antibodies produced were characterized through their ability to recognize the corresponding antigens in ELISA, Western blotting, and immunochemistry assays in rotavirus infected cells. The amount and purity of recombinant proteins obtained in this work, and the antibodies against them, are expected to be useful for the characterization of the virus-cell interaction.
Key words: rotavirus, VP5*, VP8*, recombinant proteins, E. coli BL21(DE3)
Introducción
Los rotavirus, pertenecientes a la familia Reoviridae, son los principales agentes causales de diarrea aguda y deshidratante que afecta a los infantes menores de 5 años y animales jóvenes (Esposito et al., 2011). Su partícula viral está formada por tres capas proteicas: la capa más externa compuesta por las proteínas estructurales VP4 y VP7, la capa media formada por la proteína VP6 y la capa interna formada por VP2, la cual encierra las proteínas VP1 y VP3 y el genoma viral compuesto por 11 segmentos de RNA de cadena doble (dsRNA), los cuales codifican las seis proteínas estructurales y seis proteínas no estructurales (NSP1 - NPS6) involucradas en el ciclo viral del rotavirus, incluyendo la replicación, transcripción, traducción y formación de nuevas partículas infecciosas (Estes et al., 2007). La eficiente infectividad del rotavirus depende de que la proteína VP4 sea enzimáticamente fragmentada en VP8* y VP5*.
Actualmente, se han identificado mediante estudios in vitro en las líneas celulares MA104 (línea celular epitelial de riñón de mico) y Caco-2 (línea celular de un adenocarcinoma humano) o enterocitos de vellosidades aisladas de intestino delgado (Santana et al., 2013), diferentes moléculas celulares que están involucradas en la unión y penetración del virus y que actúan como receptores de adhesión e internalización, entre las cuales se encuentran: ácido siálico (AS), integrinas α2β1, α4β1, αvβ3 y αx β2 (revisado en Acosta et al., 2009), la proteína de choque térmico Hsc70 (Gualtero et al., 2007) y la proteína disulfuro isomerasa, PDI (Calderon et al., 2012, Santana et al., 2013). Sin embargo no se ha establecido completamente con cuál de las proteínas estructurales del virus interacciona cada una de las proteínas celulares. Por esta razón, la disponibilidad de proteínas estructurales del rotavirus purificadas y la obtención de anticuerpos policlonales contra estas proteínas se convierte en una importante herramienta experimental para el desarrollo y culminación de proyectos de investigación que intentan entender los diversos mecanismos de unión y penetración del virus en sistemas de células susceptibles a la infección.
Los sistemas de expresión de proteínas se han utilizado ampliamente con diferentes propósitos en la investigación básica y en las aplicaciones biotecnológicas. Los denominados vectores de expresión (revisado en Sorensen et al., 2005; Sahdev et al., 2008) han sido de gran utilidad en este propósito al permitir la captación del material genético foráneo y la subsecuente transcripción del gen de interés seguida de su traducción en el ambiente bacteriano. La expresión de proteínas heterólogas comúnmente resulta en su acumulación en forma insoluble, dando origen a cuerpos de inclusión cuya solubilización implica tratamientos con detergentes, seguido de procedimientos dirigidos a conseguir la renaturalización de la proteína foránea (revisado en Sahdev et al., 2008). E. coli es el organismo más utilizado para expresión heteróloga de proteínas debido a sus ventajas ampliamente documentadas (revisado en Chou 2007; Sorensen et al., 2005). En el caso de proteínas de rotavirus, VP5* y VP8* han sido expresadas utilizando vectores de expresión plasmídicos (Monnier et al., 2006; Nava et al., 2004; Favacho et al., 2006) y virales (Yoder et al., 2006; Dormitzer et al., 2001). Sin embargo, la solubilidad o insolubilidad de estas proteínas en sistemas heterólogos ha dependido del sistema de expresión utilizado (Kovacs et al., 2001; Favacho et al., 2006).
En el presente trabajo, se estandarizaron protocolos para la expresión de proteínas estructurales de rotavirus con actividad antigénica utilizando un sistema de transfección bacteriana para las proteínas VP5* y VP8*. También se caracterizaron anticuerpos policlonales contra estas proteínas, para el apoyo y desarrollo de otros proyectos de investigación en pruebas de identificación mediante ELISA, "Western blotting", inmunofluoresecencia e inmunocitoquímica en ensayos de inhibición de la infección.
Metodología
Plásmidos recombinantes y transfección de bacterias
Los plásmidos pGEX-4T-VP5* y pGEX-4T-VP8* (Pharmacia, Estocolmo, Suecia) y la cepa bacteriana de E. coli BL21(DE3) (Novagen, USA) fueron donados por el laboratorio del Dr Carlos Arias (Universidad Nacional Autónoma de México, UNAM, México). El plásmido pGEX-4T-VP8* contenía la secuencia génica que comprende los nucleótidos 1 a 750 del gen que codifica la proteína rotaviral VP4 de la cepa RRV; mientras que el constructo pGEX-4T-VP5* contenía los nucleótidos 749 a 2347 de la misma cepa (Zárate et al., 2000, Pavel et al., 1997).
Las células se hicieron competentes usando una modificación del protocolo de Chung C. T., et al. (1989). Una colonia de bacterias BL21(DE3) fue inoculada en 100 ml de medio LB y cultivada a 37 ºC con agitación constante a 150 rpm durante toda la noche. Posteriormente, 1 ml del cultivo anterior se inoculó en 100 ml de medio LB hasta alcanzar una densidad óptica (OD) de 0.3 - 0.4 a 600 nm. El cultivo fue centrifugado a 5000 rpm durante 10 min y el pellet fue resuspendido en 5 ml (1/20 respecto al volumen inicial de medio LB) de medio de congelación que contenía polietilenglicol (PEG) 3350 al 10%, dimetilsulfóxido (DMSO) al 5%, NaCl 100 mM, MgCl2 10 mM, MgSO4 10 mM y glicerol al 10% disuelto en medio LB al 60% v/v en H2O estéril, pH 6,7 - 7,0, y se incubó en hielo durante 5 min. Las alícuotas fueron almacenadas en nitrógeno líquido.
Una de las alícuotas anteriores se mezcló con aproximadamente 10 ng de DNA plasmídico (pGEX-4T-VP5* o pGEX-4T-VP8*) cuantificado a 260 nm en un GeneSpec I y disuelto en 100 µl de una solución de KCl 100 mM, CaCl2 30 mM y MgCl2 50 mM. La mezcla de bacterias y DNA se agitó suavemente y posteriormente se incubó en hielo durante 20 min. Las células fueron expuestas a choque térmico a 42 ºC durante 2 min para garantizar la entrada del vector a la célula. Las células transformadas se sembraron en placas LB - agar con 100 µg/ml de ampicilina y se incubaron durante 14 h a 37 ºC.
Expresión de las proteínas virales rVP5* y rVP8*
Las alícuotas congeladas de las cepas transformadas fueron sembradas en una caja de Petri en presencia de 100 µg/ml de ampicilina y se incubaron a 37 ºC durante toda la noche. Una colonia de cada cultivo fue sembrada en 25 ml de medio líquido LB en presencia de 100 µg/ml de ampicilina y se incubaron a 37 ºC con agitación constante a 150 rpm durante 12 h. Posteriormente, se adicionaron 5 ml del cultivo anterior a 500 ml de medio LB (pGEX-4T-VP8*) o 2XYT (pGEX-4T-VP5*) y se incubaron a 37 ºC con agitación constante a 150 rpm durante 1 - 8 h hasta alcanzar una OD correspondiente a la fase exponencial del crecimiento bacteriano. A los cultivos se les adicionó el inductor isopropil β-D-tioglactopiranósido (IPTG) a una concentración final entre 0.5 - 2 mM y se incubaron durante 2, 4, 6 y 8 h a 37 ºC. Para la expresión de la proteína viral rVP5* fue necesario la adición de 2% de glucosa durante todo el procedimiento de crecimiento celular. Al finalizar el tiempo de inducción, el cultivo líquido se centrifugó a 5000 rpm durante 10 min, el pellet de células fue lavado dos veces con buffer PBS y tratado con 100 µg/ml de lisozima y se incubó durante 15 min en hielo con agitación constante. A fin de completar la lisis celular, la mezcla fue sonicada con una amplitud de 30 % durante 5 ciclos de 30 segundos, se centrifugó a 10000 rpm durante 10 min a 4 ºC y se separó el sobrenadante (fracción soluble) del pellet (fracción insoluble). El pellet fue tratado de manera consecutiva con 5 ml de urea 1, 2, 4 y 8 M en PBS a 4 ºC con agitación constante durante 2 h por cada tratamiento. Las proteínas del pellet solubilizadas en urea fueron centrifugadas a 12000 rpm durante 10 min después de cada tratamiento con las diferentes concentraciones del agente caotrópico. La expresión de las proteínas recombinantes fue evaluada y analizada mediante SDS- PAGE y "Western blotting" frente a un marcador de peso molecular, un control de bacterias E. Coli BL21(DE3) sin transfectar y un control de células E. Coli BL21(DE3) transfectadas pero sin inductor, para lo cual se cuantificó la concentración de proteínas totales de cada una de las fracciones a 280 nm respecto a una curva de calibración de BSA (albúmina de suero bovino).
Solubilización de las proteínas virales rVP5* y rVP8*
Para la solubilización de las proteínas rVP5* y rVP8* a partir del lisado de bacterias E. Coli BL21(DE3) transformadas, se utilizó el protocolo publicado por Frangioni et al.(1993), con algunas modificaciones. Las cepas bacterianas transformadas fuero cultivadas de manera independiente y las proteínas fueron expresadas teniendo en cuenta las condiciones estandarizadas. Finalizado el tiempo de expresión, las bacterias fueron centrifugadas a 5000 rpm por 10 min y lavadas dos veces con buffer STE (Tris - HCl 10 mM, pH 8.0, NaCl 150 mM y EDTA 1 mM). Posteriormente, el pellet de células fue resuspendido en 50 ml de buffer STE y tratado con 100 µg/ml de lizosima durante 15 min con agitación constante a 4 ºC. A la mezcla anterior, se adicionó 5 mM de DTT, 1 % de N-laurilsarcosina, y para completar la lisis celular, la mezcla fue sonicada durante 4 ciclos de 15 segundos a 20% de amplitud y centrifugada a 10000 rpm por 15 min a 4 ºC. El sobrenadante del lisado de las bacterias que expresaron la proteína viral rVP5*, fue tratado con de Tritón X-100, Tween 20, NP40, β-octilglucósido al 1% o CHAPS al 3%, de forma independiente. Para el lisado bacteriano de rVP8*, el sobrenadante fue diluido 10 veces en PBS o tratado con Triton X-100 al 3%. Los sobrenadantes obtenidos fueron analizados por SDS-PAGE al 12% utilizando tinción con azul de Coomassie y "Western blotting", como se describe más adelante, para verificar la presencia de las proteínas virales.
Purificación de las proteínas virales rVP5* y rVP8* mediante cromatografía de afinidad
Las proteínas virales VP5*-GST o VP8*-GST fueron purificadas con una resina de agarosa conjugada a glutationa (Molecular Probes). En una columna cromatográfica se cargaron 5 ml de la resina, la cual fue lavada con 10 volúmenes de columna de PBS, se equilibró con 5 volúmenes de buffer STE en presencia de DTT 5 mM y N-laurilsarcosina al 1 % y se incubó con el lisado bacteriano. Para la unión de las proteínas recombinantes virales a la resina, se estandarizó el tiempo de interacción para cada una de ellas. De esta manera, para la purificación de la proteína viral rVP5*, los lisados tratados con los detergentes fueron adicionados a la resina en la columna cromatográfica dos veces con el fin de retener la mayor cantidad de proteína. En el caso de la proteína viral rVP8*, el lisado diluido 10 veces con PBS o tratado con Tritón X 100 al 3 % se incubó toda la noche a 4 ºC con la resina puesto que presentó menor afinidad por el glutatión de la resina. Posteriormente, se realizaron varios lavados con PBS (aproximadamente 10 volúmenes de columna o hasta no evidenciar color con el reactivo de Bradford) y se procedió a la elución de cada proteína, para lo cual se empleó 10 mM de glutatión en Tris-HCl 50 mM, pH 8.0. Los eluídos fueron recolectados a un flujo de 1 ml cada 3 min para rVP5* ó 1 ml cada min para rVP8*. Finalmente, la resina fue tratada con 5 volúmenes de PBS que contenía NaCl 3 M y se lavó con 10 volúmenes de PBS. Paralelamente, una alícuota de cada una de las fracciones obtenidas en el análisis cromatográfico, las cuales incluyeron la fracción no retenida, los lavados con PBS, los eluídos y lavados con NaCl, fueron tratadas con el reactivo de Bradford con el objetivo de evidenciar la presencia de proteínas, mediante el cambió de color rojo/café a azul. Por otra parte, las fracciones fueron analizadas por SDS-PAGE al 12% utilizando tinción con azul de Coomassie y "Western blotting", como se describe más adelante.
Obtención de anticuerpos policlonales dirigidos contra las proteínas virales rVP5* y rVP8*
Las proteínas VP5*-GST y VP8*-GST purificadas fueron resupendidas en un buffer de digestión, que contenía Tris-HCl 50 mM, pH 8.0, y CaCl2 10 mM, y tratadas con trombina para retirar el péptido de GST, lo cual se realizó con el kit Thrombin CleanCleave (Sigma). Se tomaron 50 µl de la resina y se centrifugaron a 20 × g durante 2 min y se realizaron dos lavados con buffer de digestión 1X. Posteriormente, se adicionó 500 ng de la proteína viral purificada, se incubó durante 4 h a temperatura ambiente con agitación constante, se centrifugó a 500 rpm por 5 min y se separó el sobrenadante que contenía la proteína rotaviral y la fracción GST. Por último, se realizó un lavado de la resina con el buffer de digestión 1X para retirar el remanente de la proteína. Posteriormente, las fracciones que contenían tanto la proteína rotaviral como la fracción GST fueron separadas en SDS-PAGE al 12% en un único pozo de siembra y al igual que en los casos anteriores, se cortó la banda que correspondía a la proteína viral identificada por "Western blotting", la cual fue utilizada para la inmunización de los conejos.
Las bandas del gel que contenían aproximadamente 50 µg/banda de las proteínas rVP5* o rVP8* fueron cortadas en trozos pequeños y transferidas a una jeringa de 5 ml, luego se pasaron varias veces a través de agujas de diferentes calibres (No.18, 21 y 25) a fin de formar una suspensión en buffer PBS. Los conejos Nueva Zelanda de 2 - 3 meses de edad con suero pre-inmune libre de reactividad contra rotavirus, fueron inoculados con aproximadamente 750 µg de cada una de las proteínas virales administradas en tres dosis, con periodos de tiempo de 20 días entre cada inmunización. Las dosis fueron aplicadas vía subcutánea, distribuidas paralelamente a la columna vertebral. Después de terminado el periodo de inoculación, los conejos fueron sangrados completamente 20 días después de la tercera inmunización. Las muestras de sangre fueron incubadas a 37 °C por una hora, posteriormente a 4 ºC toda la noche, centrifugadas a 5000 rpm durante 10 min y se recuperó el suero, el cual contenía los anticuerpos contra cada una de las proteínas rotavirales. El suero fue diluido con glicerol en una proporción 1:1, dispensado en tubos de 1.5 ml estériles y almacenados a - 20 ºC.
Caracterización de los anticuerpos policlonales dirigidos contra las proteínas virales rVP5* y rVP8*
Los anticuerpos obtenidos contra las proteínas virales rVP5* y rVP8* fueron caracterizados mediante "Western blotting", ELISA e inmunocitoquímica.
"Western blotting". Diferentes cantidades de las proteínas purificadas (500 - 62.5 ng) y lisados bacterianos (100 - 25 ng) que contenían rVP5* y rVP8* fueron separados en SDS-PAGE al 12% y transferidos a membranas de PVDF a 50 mAmp durante 2 h en una cámara Mini Trans-Blot Cell (Bio- Rad Labs, Hercules, USA) con buffer de transferencia que contenía glicina 129 mM, Tris 25 mM y metanol al 10 %. Finalizada la transferencia, las membranas fueron teñidas con rojo Ponceau al 0.1 % en ácido acético al 5 %. Posteriormente, las membranas fueron lavadas con PBS para retirar la tinción y bloqueadas con leche descremada al 5% en PBS durante 2 h a 37 ºC, se lavaron con PBS dos veces y se incubaron con una dilución de 1:1000 de los anticuerpos correspondientes en PBS, Tween 20 al 0.05% y leche descremada al 0.1%. El exceso de anticuerpo no unido fue retirado mediante 5 lavados de 5 min cada uno con PBS-Tween 20 al 0.1 % y se incubaron con un anticuerpo secundario anti-conejo conjugado con peroxidasa (Santa Cruz 2030; 0.4 µg/ml) en PBS que contenía leche descremada al 0.1% y Tween 20 al 0.05 %. Se realizaron 5 lavados con PBS-Tween 20 al 0.1 % de 5 minutos cada uno para retirar el anticuerpo no unido. La detección se llevó a cabo utilizando como sustrato aminoetilcarbazol (AEC) disuelto en N-dimetilformamida. El AEC se diluyó a una concentración final de 0.268 % en buffer de acetato de sodio 100 mM, pH 5.2, se adicionó H2O2 a una concentración final de 0.03% y se filtró tres veces hasta retirar el precipitado que se forma. Finalmente, las membranas fueron lavadas con PBS. Por otra parte, se analizó el reconocimiento de las proteínas en las partículas virales, para lo cual se emplearon 500 ng de TLPs (partículas de triple capa) de RRV, pero a diferencia del ensayo anterior, se variaron las diluciones de los anticuerpos policlonales, entre 1:250 a 1:1000.
Inmunocitoquímica. Se cultivaron células MA104 en cajas de 96 pozos hasta una confluencia del 100%. La monocapa de células fue lavada dos veces con medio de cultivo e infectada con 50 µl de TLPs de RRV purificadas en una dilución 1:3200 que tenían un título de 1.8 x 108 FFU/ml, previamente tratadas con 10 µg/ml de tripsina durante 30 min a 37 ºC. Las células fueron incubadas a 37 ºC durante 1 h, lavadas dos veces e incubadas adicionalmente con 100 µl de medio MEM a 37 ºC durante 12 h. Finalizado el tiempo de infección, la monocapa fue lavada dos veces con PBS, fijada con 50 µl de metanol frío y refrigeradas a 4 ºC durante 40 min. Las células fijadas fueron incubadas de manera separada con diferentes diluciones de los anticuerpos policlonales, entre 1:250 a 1:2000 en PBS e incubadas a 37 °C durante 1 h. La monocapa celular fue lavada 5 veces con PBS-Tween 20 al 0.05%, incubada con un anticuerpo anti-conejo conjugado con peroxidasa (Santa Cruz 2030; 0.133 µg/ml) en PBS durante 45 min a 37 ºC y lavadas cinco veces con PBS-Tween 20 al 0.05%. El proceso de detección de la reacción se realizó con AEC, como se describió en el procedimiento para "Western blotting". Por último, las células fueron lavadas con agua y se determinó el porcentaje de infección en cada uno de los pozos respecto al control positivo (anti-rotavirus).
ELISA. En una placa de ELISA de 96 pozos se incubaron concentraciones crecientes de las proteínas virales recombinantes (50 ng - 2000 ng) en PBS en un volumen final de 50 µl durante toda la noche a 4 ºC. La proteína no unida fue retirada lavando dos veces con 200 µl de PBS durante cada lavado, la caja fue bloqueada con 200 µl de albúmina al 1% por pozo durante 2 h a 37 °C y lavada con 200 µl de PBS dos veces.
Las proteínas adheridas fueron incubadas durante 2 h a 37 °C con los anticuerpos policlonales respectivos en una dilución 1:1000 en PBS-Tween 20 al 0.05% que contenía albúmina al 0,.1%. El anticuerpo no unido fue retirado mediante el lavado de los pozos con 200 µl de PBS-Tween 20 al 0.1 % tres veces, dejando 5 min entre cada lavado. A continuación, se adicionó a cada pozo un anticuerpo anti-conejo conjugado a peroxidasa (Santa Cruz 2030; 0.133 µg/ml) en PBS-Tween 20 al 0.05% que contenía BSA al 0.1% y se incubó durante 1 h a 37 ºC. El exceso de anticuerpo secundario fue retirado con tres lavados de 200 µl de PBS Tween 20 al 0.1%. Finalmente, la detección se llevó a cabo mediante la adición de 50 µl por pozo de una solución de 1 mg /ml de orto-fenilendiamina (OPD) disuelta en H2O2 al 0.03 % y buffer fosfato/citrato, pH 5.0, que contienía 25 mM de ácido cítrico y 50 mM de Na2HPO4. Cuando se evidenció la formación de un color amarillo intenso, la reacción se detuvo mediante la adición de 50 µl de H2SO4 2.5 M. Como controles negativos de la técnica, se realizaron las incubaciones antes mencionadas pero en ausencia de alguno de los componentes, en cuyo lugar se adicionaron 50 µl de albúmina al 1%. Los ensayos fueron realizados por duplicado y el procedimiento se realizó dos veces de manera independiente. Para las gráficas se tabuló el Δ de densidad óptica (OD), el cual resulta de la diferencia de la absorbancia obtenida para los experimentos menos la absorbancia de los controles negativos.
Resultados
Expresión de las proteínas virales rVP5* y rVP8*
Las bacterias E. coli de la cepa BL21(DE3), deficientes en la expresión de proteasas, fueron transformadas con los plásmidos pGEX-4T-VP5* y pGEX-4T-VP8* utilizando PEG e iones metálicos mono y divalentes (Chung et al., 1989), los cuales facilitan la entrada de moléculas de DNA circular al modificar la membrana bacteriana haciéndola más permeable a estas moléculas. Para la expresión de las proteínas rVP5* o rVP8* de rotavirus fue necesario establecer las condiciones óptimas para obtener la mayor expresión de la proteína de interés. Las variables analizadas fueron: número de bacterias antes de inducir la expresión, tiempo de inducción, concentración del inductor, medio de crecimiento y presencia de cuerpos de inclusión. La expresión de las proteínas recombinantes fue determinada mediante SDS-PAGE y "Western blotting" a partir de los lisados bacterianos.
Expresión de la proteína viral rVP8*
Para la expresión de la proteína viral estructural rVP8* de rotavirus, la primera variable que se tuvo en cuenta fue el número de bacterias mediante la medición de la densidad óptica a 600 nm (OD 600 nm) por unidad de tiempo. Así, se identificaron las diferentes fases del crecimiento microbiano (latencia, exponencial y estacionaria). Se realizó una curva de crecimiento de las bacterias transformadas, para lo cual se determinó la OD 600 nm cada hora, teniendo en cuenta la relación directa entre el incremento del número de células y el valor de absorbancia registrado sin discriminar la viabilidad de las mismas.
A partir de esta curva de crecimiento se pudo definir la fase de latencia, comprendida entre 0 - 1 h, la fase exponencial, entre 1 - 4 h y por último, la fase estacionaria, a partir de las 4 horas de crecimiento. De esta manera, se decidió evaluar la expresión de la proteína viral rVP8* en rangos de absorbancia entre 0.1 - 0.6 (en la curva se graficó el log (OD x 100) vs tiempo) que corresponden a la fase exponencial y el inicio de la fase estacionaria del crecimiento bacteriano (figura 1a).
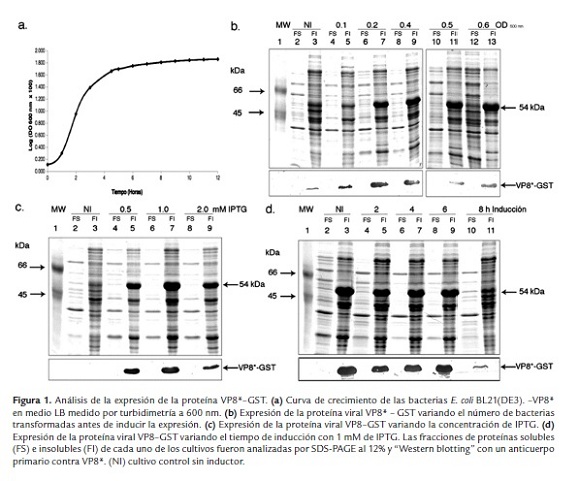
Los carriles 2 y 3 del análisis de SDS-PAGE (figura 1b) corresponden a las proteínas de las fracciones soluble e insoluble del cultivo bacteriano que no fue tratado con IPTG, respectivamente, en los cuales se observó una baja expresión basal de la proteína viral en la fracción de proteínas insolubles alrededor de 54 KDa. En los carriles 4, 6, 8, 10 y 12, que corresponden a las fracciones solubles de los lisados bacterianos que fueron tratados con IPTG una vez habían alcanzadas las OD 600 nm de 0.1, 0.2, 0.4, 0.5 y 0.6, respectivamente, no se observó la presencia de la proteína recombinante. En los carriles 5, 7, 9, 11 y 13 se sembraron las fracciones insolubles de los lisados bacterianos anteriores y se observó que a medida que se incrementaba el número de bacterias, la cantidad de rVP8* era mayor. De esta manera, la expresión de la proteína viral rVP8* se localizó en la fracción insoluble, independientemente de la OD a la cual se realizó la inducción de la expresión de la proteína viral.
Se cuantificaron las proteínas totales a 280 nm y se analizaron por SDS-PAGE al 12%, 100 µg/pozo de cada fracción proteica. La banda correspondiente a la proteína rVP8* fue comparada visualmente con una curva de calibración de BSA analizada en el mismo gel teñido con azul de Coomassie y su proporción (porcentaje) determinada con relación a la proteína total cargada en el respectivo pozo del gel. En la electroforesis se observó que la mayor proporción de proteína viral (1.59% respecto a la proteína total) se obtuvo cuando la inducción se realizó durante 4 horas a partir de un cultivo que había alcanzado una OD600 nm de 0.5.
Otros factores que afectaron la cantidad y solubilidad de las proteínas recombinantes durante su expresión, fueron la concentración del inductor y el tiempo de inducción. Se evaluaron diferentes concentraciones de IPTG, entre 0.5 y 2 mM (figura 1c), y se observó que la mayor proporción de proteína viral (1.99% respecto a la proteína total) se obtuvo cuando el cultivo bacteriano fue tratado con 1 mM de IPTG, localizándose la proteína en la fracción insoluble del lisado bacteriano (figura 1c, carril 7). A una menor o mayor concentración del inductor, la cantidad de proteína viral expresada fue menor (figura 1c, carriles 5 y 9). Por otra parte, las fracciones de proteínas solubles correspondientes no presentaron la proteína recombinante (figura 1c, carriles 4, 6 y 8).
Igualmente, se determinó el efecto de los diferentes tiempos de inducción, entre 2 y 8 h, con 1 mM de IPTG aplicado al cultivo cuando éste alcanzó una OD 600 nm de 0.5 (figura 1d). En las fracciones insolubles, correspondientes a los lisados bacterianos que fueron tratados durante 2, 4, 6 y 8 horas con 1mM de IPTG (carriles 5, 7, 9 y 11, respectivamente), se observó la presencia de la proteína viral, correspondiente a la banda más ancha, identificada mediante "Western blotting". En las fracciones solubles correspondientes (carriles 4, 6, 8 y 10) no se evidenció la presencia de la proteína viral. Se determinó que la concentración de la proteína viral VP8*-GST fue mayor (1.99 % respecto a la proteína total) cuando el cultivo bacteriano transformado fue inducido durante 6 a 8 h, respecto a la inducción de 2 y 4 h (0.79 % - 0.92 % respecto a la proteína total).
Teniendo en cuenta que la expresión de la proteína viral rVP8* se localizó en la fracción de proteínas insolubles, se realizó el crecimiento bacteriano y la expresión a una temperatura de 25 ºC. Se observó que a pesar de disminuir la temperatura durante la inducción de la proteína recombinante, ésta se siguió localizando en la fracción insoluble y su rendimiento fue menor (datos no mostrados). En resumen, las bacterias transformadas con el vector pGEX-4T-VP8* fueron cultivadas hasta alcanzar una OD 600 nm de 0.5 al momento de inducir la expresión de la proteína con 1 mM de IPTG durante 6 h a 37 ºC.
Expresión de la proteína viral rVP5*
Para la expresión de la proteína viral estructural rVP5* se emplearon las condiciones previamente estandarizadas para la expresión de la proteína viral VP8*-GST; sin embargo, la cantidad de proteína expresada fue muy baja en comparación con los resultados obtenidos para rVP8*. Se aumentó la expresión de la proteína recombinante variando el medio de cultivo y adicionando glucosa durante el crecimiento bacteriano.
Se analizó la expresión de la proteína VP5* - GST creciendo las bacterias transformadas en dos medios diferentes: LB y 2XYT. El primero tiene los nutrientes esenciales básicos y el segundo es un medio enriquecido, con una mayor concentración de triptona y extracto de levadura, respecto a LB. La presencia de la proteína viral fue identificada en la fracción de proteínas insolubles mediante "Western blotting" con un anticuerpo anti-VP5* y anti-GST y presentó una masa molecular de 75 KDa (figura 2c). Cuando las bacterias fueron cultivadas en medio LB sin inductor, no hubo expresión basal de rVP5* (carril 2); sin embargo, cuando el cultivo se realizó en medio 2XYT, se incrementó la expresión basal de la misma (carril 3), pero en presencia de glucosa al 2% esta expresión se inhibió (carril 4). La mayor proporción de proteína viral (2.38% respecto a la proteína total) se obtuvo cuando el cultivo se realizó en medio 2XYT en presencia de glucosa al 2%, con 1 mM de IPTG (carril 8), respecto a las bacterias que fueron cultivadas en este mismo medio pero en ausencia de glucosa (1.98 % respecto a la proteína total) (carril 7) y en medio LB con glucosa (1.36 % respecto a la proteína total) y sin glucosa en presencia del inductor (1.29 % respecto a la proteína total) (carril 5 y 6).
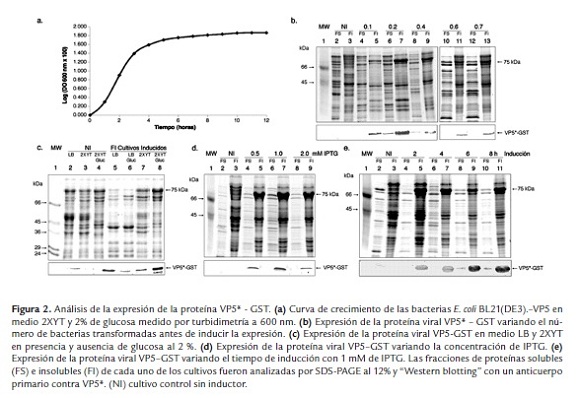
Con el fin de incrementar la proporción de rVP5* respecto a la proteína total e intentar obtenerla en su forma soluble, se analizó la expresión de la proteína recombinante cuando la inducción se realizó a diferentes OD 600 nm al momento de adicionar IPTG. Para esto se realizó la curva de crecimiento para las bacterias BL21(DE3) transfectadas con el plásmido pGEX-4T-VP5* con el fin de identificar la fase de crecimiento exponencial y estacionaria de las bacterias transformadas en medio 2XYT y en presencia de glucosa al 2% (figura 2a). Se encontraron los mismos resultados obtenidos en el cultivo de bacterias transformadas con el vector pGEX-4T-VP8*, con respecto al tiempo de cultivo requerido para alcanzar las diferentes etapas del crecimiento bacteriano.
Después de identificado el tiempo en el cual se llevó a cabo la fase de crecimiento exponencial y estacionaria de las bacterias transformadas, fue posible evaluar la expresión de la proteína viral cuando los cultivos alcanzaron las diferentes OD 600 nm (0.1 - 0.7) al momento de adicionar el inductor. No se evidenció expresión basal de la proteína en las fracciones soluble e insoluble del lisado bacteriano que no fue inducido con IPTG, carriles 2 y 3 (figura 2b). Tampoco se detectó en las fracciones solubles de lisados bacterianos que fueron tratados con el inductor una vez el cultivo alcanzó una OD 600 nm de 0.1, 0.2, 0.4, 0.6 y 0.7, como se observa en los carriles 4, 6, 8, 10 y 12, respectivamente. Se identificó la presencia de rVP5* en las fracciones insolubles (carriles 5, 7, 9, 11 y 13). Esto se observó a pesar de variar el número de bacterias antes de estimular su expresión. La mayor proporción de la proteína recombinante (3.20 % respecto a la proteína total) se obtuvo cuando el cultivo alcanzó una OD 600 nm entre 0.2 - 0.4 antes de realizar la inducción con 1 mM de IPTG durante 4h.
Otra variable que afecta la expresión de una proteína recombinante es la concentración del inductor (figura 2d). La proteína viral rVP5* se mantuvo en la fracción de proteínas insolubles (carriles 5, 7 y 9), aún cuando se varió la concentración del inductor. No se observó la presencia de la proteína viral en las fracciones solubles de los lisados anteriores (carriles 4, 6 y 8). Se determinó que la mayor proporción de la proteína (3.78% respecto a la proteína total) fue obtenida cuando al cultivo se le adicionó 0.5 y 1 mM de IPTG. En concentraciones de 2 mM del inductor disminuyó la proporción de la proteína viral (0.80% respecto a la proteína total).
Por otra parte, se evaluó el tiempo de inducción. Como se observa en la figura 2e, la banda correspondiente a la proteína rVP5* fue detectada solo en las fracciones insolubles de los lisados bacterianos inducidos durante 2, 4, 6 y 8 h (carriles 5, 7, 9 y 11). No se observó en las fracciones solubles, carriles 4, 6, 8 y 10. La mayor concentración de rVP5* (3.82% respecto a la concentración total de proteínas) fue obtenida cuando el cultivo fue inducido durante 4 h. Sin embargo, no se observaron cambios drásticos en la cantidad de proteína expresada en los diferentes tiempos analizados.
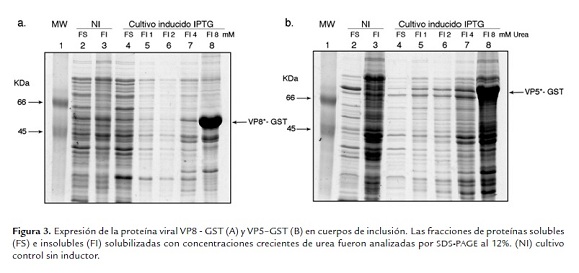
En resumen, las bacterias transformadas con el vector pGEX-4T-VP5* fueron cultivadas hasta alcanzar una OD 600 nm de 0.2 - 0.4 al momento de inducir la expresión de la proteína con 0.5 mM de IPTG durante 4 h a 37 ºC. La expresión de VP8* o VP5* fue localizada en la fracción de proteínas insolubles en las diferentes condiciones analizadas, lo cual sugiere la formación de los denominados cuerpos de inclusión. El pellet obtenido de las proteínas recombinantes VP8*-GST y VP5*-GST fue tratado con concentraciones crecientes de urea (1 - 8M), y la solubilidad se alcanzó cuando se empleó 8 M de urea (figura 3a y b, carril 8), sugiriendo la formación de cuerpos de inclusión.
Purificación de las proteínas virales rVP8* y rVP5*
Para la purificación de la proteína rVP8* se empleó la metodología descrita por Frangioni et al. (1993), utilizando N-laurilsarcosina y DTT para lisar bacterias y solubilizar los cuerpos de inclusión. Adicionalmente, se empleó Tritón X-100 al 3% como agente renaturante; sin embargo, la cantidad de proteína recombinante purificada fue muy poca respecto a la cantidad de proteína total identificada en el lisado bacteriano (figura 4a, carril 4). Otra técnica de renaturación consistió en diluir el lisado bacteriano con PBS. De esta manera, la cantidad de rVP8* recuperada y el número de fracciones eluídas que contenían la proteína recombinante (figura 4b, carril 5 - 7) fue mayor respecto a los demás ensayos, partiendo del mismo volumen de muestra.
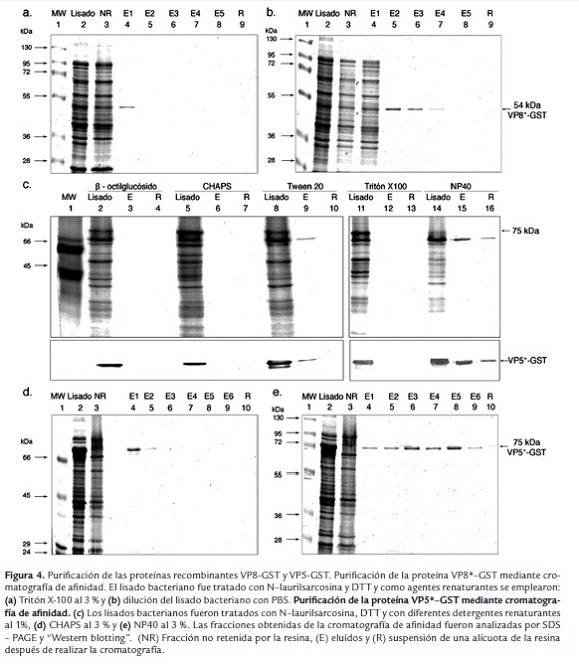
Para la purificación de rVP5* mediante cromatografía de afindiad, los lisados bacterianos fueron tratados con los siguientes detergentes antes de adicionarlos a la resina: Tritón X-100, Tween 20, NP40, octilglucósido o CHAPS. Los detergentes que incrementaron la afinidad de la proteína recombinante por el glutatión de la resina fueron CHAPS y NP40 al 1 % (figura 4c, carril 9 y 15); sin embargo, el rendimiento de la purificación fue muy bajo teniendo en cuenta la cantidad de proteína purificada respecto a la cantidad total en el lisado bacteriano (figura 4c, carril 8 y 14). Con los detergentes, β-octilglucósido, Tween 20 y Tritón X100, no se logró retener la proteína recombinante (carriles 3, 6 y 12). Con el objetivo de incrementar la cantidad de proteína purificada, se aumentaron las concentraciones de los detergentes. El tratamiento con CHAPS al 3 % permitió la formación de una suspensión que fue retirada, centrifugada y analizada por electroforesis y "Western blotting", identificándose la proteína viral junto con otras proteínas bacterianas. La proteína que se mantuvo soluble se adicionó a la resina, pero se recuperó una cantidad baja respecto a la rVP5* encontrada en el lisado total (figura 4d, carril 4 y 5). Con NP40 al 3 % como agente renaturante, se logró purificar la mayor cantidad de proteína VP5* - GST en comparación con CHAPS al 3 %, teniendo en cuenta que se partió del mismo lisado bacteriano (figura 4d, carril 4 a 10). Para disminuir la concentración del detergente y emplear las proteínas en ensayos de interacción en condiciones acelulares y celulares, las fracciones eluídas se diluyeron con PBS y fueron concentradas mediante un Amicon Ultra - 4 (Millipore).
Obtención de sueros hiperinmunes anti-rVP8* y rVP5*
Las proteínas purificadas VP8*-GST o VP5*-GST fueron tratadas con el kit Thrombin CleanCleave (Sigma) para retirar el polipéptido GST (figura 5a y b) y las fracciones proteícas correspondientes a la proteína viral fueron utilizadas como antígeno para la inoculación en conejos.
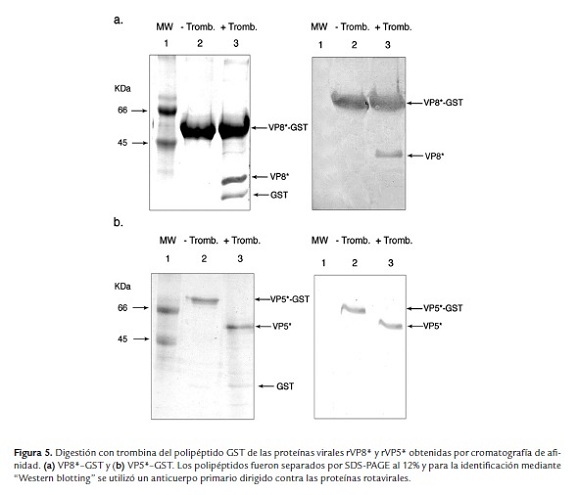
El suero hiperinmune anti-rVP5* reconoce en el "Western blotting" una banda de aproximadamente 75 KDa en los lisados bacterianos transformados con el vector pGEX-4T-VP5* (figura 6a). Sin embargo, en altas concentraciones del lisado se observaron bandas adicionales (figura 6a). El suero hiperinmune anti-rVP5*, en una dilución 1:1000, reconoce la proteína purificada a partir de 62.5 ng/pozo (figura 6c), con buen reconocimiento a 250 y 500 ng/pozo (figura 7c, carril 3 y 4). También se observó una banda de menor peso molecular, posiblemente un producto de degradación de la proteína recombinante o a un reconocimiento inespecífico del anticuerpo.
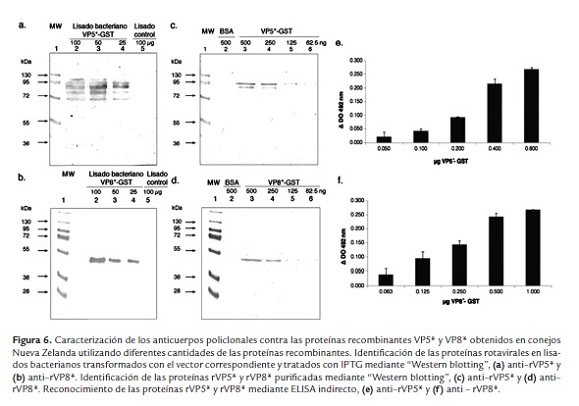
El suero hiperinmune anti-rVP8* reconoció solamente una banda en los lisados bacterianos (figura 6b) y no se observaron bandas inespecíficas. El suero hiperinmune anti-rVP8*, en una dilución 1:1000, reconoce la proteína purificada a partir de 62.5 ng/pozo, con buen reconocimiento a 250 y 500 ng/pozo (figura 6d, carril 3 y 4). Anti-rVP5* o rVP8* no identificaron la proteína BSA utilizada como control (figura 6c y d, carril 1).
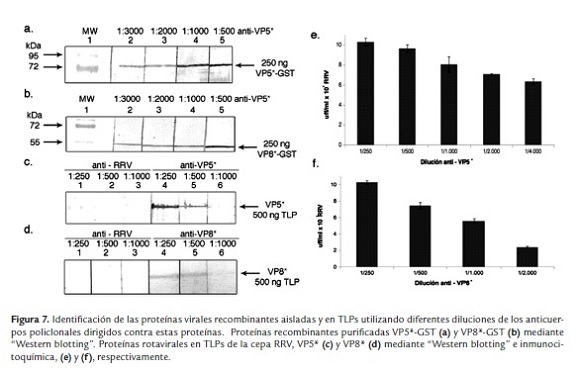
Para el ensayo de ELISA, se observó un incremento en la DO directamente proporcional al aumentar la cantidad de proteína (figura 6e - f). Para identificar la dilución límite de reconocimiento de antígeno en "Western blotting" se analizaron en electroforesis 250 ng/pozo de las proteínas purificadas rVP5* o rVP8* y transferidas a membranas de PVDF. Las diluciones de los anticuerpos fueron entre 1:500 a 1:3000. Se observó buen reconocimiento con diluciones 1:3000 (figura 7a y b, carril 2). Utilizando "Western blotting" se quiso determinar si los sueros reconocen las proteínas del virión. Para esto, se emplearon 500 ng/pozo de TLPs y se utilizaron diferentes diluciones de los sueros anti-rVP8* o rVP5* (1:250 a 1:1000) comparado con un suero hiperinmune anti-rotavirus que reconoce todas las proteínas estructurales del virus, generado en el laboratorio. Se observó reconocimiento por los sueros anti-rVP5* y rVP8*, en una dilución 1:250 y 1:500, de las proteínas en las TLPs (figura 7c y d, carril 4 y 5). La dilución 1:1000 no mostró reconocimiento (figura 7c y d, carril 6). Los sueros anti rVP8* o rVP5* reconocieron mejor las proteínas virales de las TLPs que el suero anti-rotavirus generado contra todas las proteínas estructurales (figura 7c y d, carril 1, 2 y 3).
Para el ensayo de inmunocitoquímica, células MA104 infectadas con TLPs del rotavirus RRV se incubaron con diluciones de suero anti-rVP5* o rVP8*. Anti-rVP5* reconoció las células que son positivas al antígeno en las diluciones 1:250- 1:4000 a partir de las cuales se estimó un título viral correspondiente a 6.34 x 107 uff/ml (figura 7e), mientras que anti-rVP8* reconoció las células que son positivas al antígeno respectivo en las diluciones 1:250- 1:2000, de tal manera que la proporción de células positivas correspondió a un título viral de 5.56 x 107 uff/ml con la dilución 1:1000 (figura 7f).
Discusión
La mayor expresión de rVP8* no dependió de células que se encuentren en duplicación constante, no fue afectada por la concentración de nutrientes o productos de desecho presentes en el medio ni por el tiempo de inducción sugiriendo que su expresión es constante y no depende del estado metabólico de la célula. Por otra parte, concentraciones superiores a 1 mM de IPTG disminuyeron la concentración de la proteína recombinante, probablemente debido a que se disminuye la estabilidad del transcrito y rRNA, aún cuando los niveles de mRNA se pueden ver ligeramente incrementados (Wood et al., 1991; Jinks et al., 1983; Casadaban et al., 1983; Gausing, 1977; Nilsson et al., 1987).
La expresión basal (en ausencia de inductor) de la proteína viral rVP5*, se incrementó en las bacterias cultivadas en medio 2XYT, el cual contiene una mayor concentración de triptona y extracto de levadura. Este comportamiento se controló empleando glucosa al 2%, la cual disminuye los niveles de cAMP intracelular que son requeridos para activar la expresión de la RNA polimerasa T7 después de su interacción con la proteína de unión a cAMP (CAP) (Baneyx, 1999; Kuo et al., 2003). En presencia del inductor se observó una mayor proporción de la proteína viral en medio 2XYT con glucosa en comparación con el cultivo realizado en medio LB. Se debe destacar que no se evidenció un cambio significativo en la proporción de la expresión de rVP5* a los diferentes tiempos de inducción evaluados. La disminución en la proporción de rVP5* expresada a las 6 y 8 h de inducción, podría atribuirse a su degradación; sin embargo, no se evidenciaron bandas reactivas de rVP5* de menor peso molecular en "Western blotting".
Las proteínas rVP8* y rVP5* se localizaron en la fracción denominada insoluble y cuya solubilidad se logró cuando se utilizaron concentraciones de urea de 8 M. Estas agregaciones proteícas se denominan cuerpos de inclusión y se forman en el espacio citoplasmático y periplásmico de las células de E. coli cuando se obtienen altos niveles de expresión de proteínas heterólogas (más del 2% del total de proteínas bacterianas) o cuando la proteína expresada es hidrofóbica. Estas agregaciones son principalmente formadas por interacciones hidrofóbicas no nativas entre estructuras con plegamiento intermedio, en las cuales los dominios hidrofóbicos se encuentran expuestos (Mohan et al., 2005; Maeda et al., 1995; Ejima et al., 1999).
Por otra parte, en diferentes estudios se ha expresado la proteína viral VP8* mediante varios sistemas, entre ellos: infección de células de insecto Sf9 con un baculovirus recombinante (Mackow et al., 1989; Flore et al., 1991; Gorzigilia et al., 1990; Khodabandehloo et al., 2009), inducción de la expresión en bacterias E. coli JM109 transfectadas con el vector pGEMEX-1-VP8* o pGEX-4T-2-VP8* (Kovacs et al., 2001; Larralde et al., 1991) y bacterias BL21(DE3) transfectadas con el vector pGEX-4T-2-VP8* o pET28a(+) (Monnier et al., 2006; Nava et al., 2004; Favacho et al., 2006). Todos los sistemas de expresión permitieron obtener la proteína viral de forma soluble, excepto cuando la expresión se realizó en bacterias BL21(DE3) con el vector pGEX-4T-2-VP8* y la inducción se llevó a cabo cuando el cultivo bacteriano alcanzó una OD 600 nm entre 0.2 - 0.4 y se indujo con 1 mM de IPTG durante 3 h a 37 ºC, datos que confirman los resultados reportados en este trabajo. Sin embargo, en un estudio posterior se demostró que la disminución en la temperatura a 25 ºC durante la inducción de la proteína, incrementa su concentración en el sobrenadante o fracción de proteínas solubles (Dormitzer et al, 2002). En este trabajo, la temperatura más baja utilizada durante el crecimiento bacteriano fue de 25 ºC, factor que no afectó la solubilización de la proteína pero si su expresión. Estos resultados muestran que la solubilidad de la proteína recombinante no solo depende de las características bioquímicas o de la concentración de la proteína en el cultivo, sino del sistema de expresión empleado (Kovacs et al., 2001; Favacho et al., 2006).
La insolubilidad de rVP5* ha sido previamente reportada en bacterias E. coli BL21(DE3) y JM109 transformadas con los vectores pET28a(+) y pGEMEX-1 recombinantes, respectivamente (Larralde et al., 1991; Dowling et al., 2000). Por otra parte, existen reportes en los cuales se demuestra que la expresión de esta proteína viral en bacterias E. coli BL21(DE3) con el vector pGEX-4T-2 puede darse en forma soluble si la inducción de la expresión se realiza a 20 ºC, con bajas concentraciones de IPTG (0.01 mM) y es co-purificada con la chaperona GroEL (Kamata et al., 1994); sin embargo, este procedimiento requiere de un paso adicional que consiste en retirar la chaperona con ATP (Rohman et al., 2000). Cabe mencionar que esta variable no fue tenida en cuenta en el presente estudio durante el proceso de optimización de la expresión de esta proteína recombinante.
La purificación de las proteínas recombinantes VP5*-GST y VP8*-GST se realizó mediante cromatografía de afinidad. Para lo cual, se empleó como técnica de solubilización N-laurilsarcosina, detergente iónico alcalino que ha sido ampliamente utilizado para solubilizar casi todas las proteínas unidas a GST y en general agregaciones proteícas (Frangioni et al., 1993; McNally et al., 1991), debido a que dispersa los cuerpos de inclusión en estructuras monomoleculares por la fuerte repulsión electrostática del complejo proteína-detergente. La concentración empleada de N-laurilsarcosina para solubilizar las proteínas bacterianas y los cuerpos de inclusión fue de 1%, superior al valor de constante micelar crítica (CMC) determinado para este detergente (0.427%), el cual permitió la solubilización de las agregaciones protéicas. Sin embargo, el uso de esta concentración del detergente aniónico afecta considerablemente la estructura de la proteína y por tanto su interacción con la resina de agarosa unida a glutatión, puesto que depende básicamente de la estructura tridimensional del polipéptido GST.
Estudios previos han demostrado que detergentes no iónicos pueden formar micelas mezcladas con el detergente iónico y previenen su precipitación en presencia de iones divalentes, como calcio y magnesio (Tsumoto et al., 2010; Stewart et al., 1991), y no afectan la interacción de las proteínas unidas a GST a la resina de agarosa (Smith et al., 1988). Por esta razón, fue necesario utilizar un detergente no iónico que permitiera el replegamiento de las proteínas presentes en el lisado bacteriano.
El detergente no iónico utilizado en el proceso de purificación de la proteína viral rVP8* fue Tritón X-100 (Frangioni et al., 1993) y se observó la unión de la proteína recombinante a la resina; sin embargo, la cantidad de proteína retenida fue muy baja en comparación con el total de la proteína recombinante presente en el lisado bacteriano total y la encontrada en la fracción no retenida, obtenida después de la interacción del lisado con la resina. Por lo tanto, se empleó una estrategia experimental diferente para inducir el replegamiento del polipéptido que consistió en diluir 10 veces con buffer PBS el lisado celular solubilizado con N-laurilsarcosina al 1% (Wall et al, 1995). Este tratamiento resultó más eficiente, puesto que se logró obtener un mayor número de eluídos de la proteína VP8*-GST.
Para la purificación de la proteína VP5*-GST, se emplearon como agentes renaturantes detergentes no iónicos (Tween 20, NP40 y β-octilglucósido) y un detergente zwiteriónico (CHAPS) (Lundbäck et al., 2003). Estos detergentes fueron utilizados porque la solubilización de las agregaciones proteicas, mediante el uso de N-laurilsarcosina, encapsulan las proteínas y las disgregan; mientras que los detergentes no iónicos o zwiteriónicos forman grandes micelas o bicelas mezcladas que incorporan las moléculas del detergente iónico (N-laurilsarcosina), disminuyendo su concentración alrededor de la proteína recombinante, dejando sitios activos libres o facilitando el replegamiento de la proteína. Por esta razón, se recomienda purificar proteínas recombinantes mediante esta metodología ensayando diferentes detergentes o mezcla de ellos, con el objeto de tener un buen rendimiento y una actividad mayor al 80% (Jinks et al., 1983; Dormitzer et al., 2001; Kamata et al., 1994; Rohman et al., 2000; Lilie et al., 1998; Burgess, 1996; Tao et al., 2010).
Los anticuerpos policlonales contra las proteínas rVP5* y rVP8* detectaron las respectivas proteínas recombinantes mediante "Western blotting", en condiciones denaturantes y ELISA, en condiciones nativas. Igualmente, estos anticuerpos reconocieron las proteínas virales expresadas en células infectadas en su condición nativa durante el proceso infeccioso y en TLPs de RRV purificadas. Estos resultados muestran que las proteínas estructurales del rotavirus rVP5* y rVP8* indujeron una eficiente respuesta inmune en los conejos que fueron inoculados. En su conjunto, las proteínas recombinantes obtenidas en el presente trabajo, y los anticuerpos inducidos por ellas, constituyen importantes herramientas para los estudios de caracterización de las proteínas virales y su interacción con moléculas de la superficie celular de la célula hospedera durante la entrada el virus.
Referencias bibliográficas
1 Acosta, O., Calderón, M. N., Moreno L. P., Guerrero, C. A. 2009. Un modelo del mecanismo de entrada de los rotavirus a la célula hospedera. Revista de la Facultad de Medicina de la Universidad Nacional. 57: 124-148. [ Links ]
2 Baneyx F. 1999. Recombinant protein expression in Escherichia coli. Current Opinion in Biotechnology. 10: 411 - 421. [ Links ]
3 Burgess R. 1996. Purification of overproduced Escherichia coli RNA polymerase sigma factors by solubilizing inclusion bodies and refolding from sarkosyl. Methods in Enzymology. 273: 145-149. [ Links ]
4 Calderon M, Guerrero CA, Acosta O, Lopez S, Arias CF. 2012. Inhibiting rotavirus infection by membrane-impermeant thiol/disulfide exchange blockers and antibodies against protein disulfide isomerase. Intervirology. 55(6): 451-464. [ Links ]
5 Casadaban M, Martinez - Arias A, Shapira S and Chou J. 1983. b-Galactosidase gene fusions for analyzing gene expression in Escherichia coli and Yeast. Methods in Enzymology. 100: 293 - 315. [ Links ]
6 Chou C. 2007. Engineering cell physiology to enance recombinant protein production in Escherichia coli. Applied Microbiology Biotechnology. 76: 521 - 532. [ Links ]
7 Chung C, Suzanne L, Niemela S and Miller R. 1989. One - step preparation of competent Escherichia coli: Transformation and storage of bacterial cells in the same solution. Proceedings of the National Academy of Science. 86: 2172 - 2175. [ Links ]
8 Dormitzer P, Greenberg H and Harrison S. 2001. Proteolysis of monomeric recombinant rotavirus VP4 yields an oligomeric VP5* core. Journal of Virology. 75: 7339 - 7350. [ Links ]
9 Dormitzer P, Sun Z, Wagner G and Harrison S. 2002. The rhesus rotavirus VP4 sialic acid binding domain has a galectin fold with a novel carbohydrate binding site. The EMBO Journal. 21: 885 - 897. [ Links ]
10 Dowling W, Denisova E, Lamonica R and Mackow E. 2000. Selective membrane permeabilization by the rotavirus VP5* protein is abrogated by mutations in an internal hydrophobic domain. Journal of Virology. 74: 6368 - 6376. [ Links ]
11 Ejima D, Watanabe M, Sato T, Date M, Yamada N and Takahara Y. 1999. High yield refolding and purification process for recombinant human Interleukin-6 expressed in Escherichia coli. Biotechnology and Bioengineering. 62: 301-310. [ Links ]
12 Esposito D, Holman RC, Haberling DL, Tate JE, Podewils LJ, Glass RI, et al. 2011. Baseline estimates of diarrhea-associated mortality among United States children before rotavirus vaccine introduction. The Pediatric Infectious Disease Journal. 30: 942-947. [ Links ]
13 Estes M, Kapikian AZ. 2007. Rotaviruses. In: Knipe DM, Howley PM, eds. Fields Virology. 5th ed. Philadelphia.1917-1974. [ Links ]
14 Favacho A, Kurtenbach E, Sardi S and Gouvea V. 2006. Cloning, expression and purification of recombinant bovine rotavirus hemagglutinin, VP8*, in Escherichia coli. Protein Expression and Purification. 46: 196 - 203. [ Links ]
15 Flore L, Greeberg H. B, Mackow E. R. 1991. The VP8 fragment of VP4 is the rhesus rotavirus hemagglutinin. Virology. 181: 553 - 563. [ Links ]
16 Frangioni J, Neel B. 1993. Solubilization and purification of enzymatically active glutathione S - transferase (pGEX) fusion proteins. Analytical Biochemistry. 210: 179 - 187. [ Links ]
17 Gausing K. 1977. Regulation of ribosome production in Escherichia coli: Synthesis and stability of ribosomal RNA and of ribosomal protein messenger RNA at different growth rates. Journal of Molecular Biology. 115: 335 - 342. [ Links ]
18 Gorzigilia M, Larralde G, Kapikian A and Chanock R. 1990. Antigenic relationships among human rotaviruses as determined by outer capsid protein VP4. Proceedings of the National Academy of Science. 87: 7155 - 7159. [ Links ]
19 Gualtero D, Guzman F, Acosta O, Guerrero CA. 2007. Amino acid domains 280-297 of VP6 and 531-554 of VP4 are implicated in heat shock cognate protein Hsc70-mediated rotavirus infection. Archives of Virology. 152: 2183-96. [ Links ]
20 Jinks-Robertson S, Gourse R and Nomura M. 1983. Expression of rRNA and tRNA genes in Escherichia coli: Evidence for feedback regulation by products of rRNA operons. Cell. 33: 865. [ Links ]
21 Kamata T and Takad Y. 1994. Direct binding of collagen to the I domain of Integrin a2b1 (VLA-2, CD49b/CD29) in a divalent cation-independent manner. The Journal of Biological Chemistry. 269: 26006 - 26010. [ Links ]
22 Khodabandehloo M, Shahrabadi M, Keyvani H and Bambai B. 2009. Cloning and expression of simian rotavirus spike protein (VP4) in insect cells by baculovirus expression system. Iranian Biomedical Journal. 13: 9-18. [ Links ]
23 Kovacs - Nolan J, Sasaki E, Yoo D and Mine Y. 2001. Cloning and expression of human rotavirus spike protein, VP8*, in Escherichia coli. Biochemical and Biophysical Research Communications. 282: 1183 - 1188. [ Links ]
24 Kuo J, Chang Y, Tseng C. 2003. Growth rate regulation of lac operon expression in Escherichia coli is cyclic AMP dependent. FEBS Letters. 553: 397 - 401. [ Links ]
25 Larralde G, Li B, Kapikian A and Gorziglia M. 1991. Serotype - specific epitope(s) present on the VP8 subunit of rotavirus VP4 protein. Journal of Virology. 65: 3213 - 3218. [ Links ]
26 Lilie H, Schwarz E and Rudolph M. 1998. Advances in refolding of proteins produced in E. coli. Current Opinion in Biotechnology. 9: 497-501. [ Links ]
27 Lundbäck A. K, Haneskog L, Andersson L, Heijbel A and Birse D. 2003. Optimal solubilization screening strategies for GST-fusion membrane proteins. Life Science News. 15: 10 - 12. [ Links ]
28 Mackow E. R, Barnett J. W, Chan H, Greenberg H. B. 1989. The rhesus rotavirus outer capsid protein VP4 functions as a hemagglutinin and is antigenically conserved when expressed by a baculovirus recombinant. Journal of Virology. 63: 1661 - 1668. [ Links ]
29 Maeda Y, Koga H, Yamada H, Ueda T and Imoto T. 1995. Effective renaturation of reduced lysozyme by gentle removal of urea. Protein Engineering Design and Selection. 8: 201- 205. [ Links ]
30 McNally E, Sohn R, Frankel S and Leinwand L. 1991. Expression of myosin and actin in Escherichia coli. Methods in Enzymology. 196: 368-389. [ Links ]
31 Mohan S, Kumar A. 2005. Solubilization and refolding of bacterial inclusion body proteins. Journal of Bioscience and Bioengineering. 99: 303 - 310. [ Links ]
32 Monnier N, Higo - Moriguchi K, Sun Z, Venkataram B, Taniguchi K and Dormitzer P. 2006. High - resolution molecular and antigen structure of the VP8* core of a sialic acid independent human rotavirus strain. Journal of Virology. 80: 1513 - 1523. [ Links ]
33 Nava P, Lopez S, Arias C, Islas S and Gonzalez L. 2004. The rotavirus surface protein modulates the gate and fence function of tight junctions in epithelial cells. Journal of Cell Science. 117: 5509 - 5519. [ Links ]
34 Nilsson G, Belasco J, Cohen S and Von Gabain A. 1987. Effect of premature termination of translation on mRNA stability depends on the site of ribosome release. Proceedings of the National Academy of Science. 84: 4890 - 4898. [ Links ]
35 Pavel I, López S, Segovia L and Arias C. 1997. Functional and structural analysis of the sialic acid-binding domain of rotaviruses. Journal of Virology. 71: 6749 - 6756. [ Links ]
36 Poncet D, Corthier G, Charpilienne A and Cohen J. 1990. A recombinant vaccinia virus expressing the major capsid protein of simian rotavirus - induced anti-rotavirus antibodies. Virus Research. 15: 267 - 274. [ Links ]
37 Rohman M and Harrison - Lavoie K. 2000. Separation of copurifying GroEL from glutathione- S - transferase fusion proteins. Protein Expression and Purification. 20: 45 - 47. [ Links ]
38 Sahdev S, Khattar S and Singh K. 2008. Production of active eukaryotic proteins through bacterial expression systems: A review of the existing biotechnology strategies. Molecular and Cellular Biochemistry. 307: 249 - 264. [ Links ]
39 Santana Y, Guerrero C and Acosta Orlando. 2013. Implication of Hsc70, PDI and integrin avb3 during the entry of rotavirus ECwt into small intestinal villi from suckling mouse. Archives of Virology. DOI 10.1007/s00705-013-1626-6 2013. [ Links ]
40 Smith D and Johnson K. 1988. Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione - S -transferase. Gene. 67: 31 - 40. [ Links ]
41 Sorensen H and Mortensen K. 2005. Soluble Expression of recombinant proteins in the cytoplasm of Escherichia coli. Microbial Cell Factories. 4: 1 - 8. [ Links ]
42 Stewart F, Regina S and Leinwand L. 1991. The Use Of Sarkosyl In Generating Soluble Protein After Bacterial Expression. Proceedings of the National Academy of Science. 88: 1192 - 1196. [ Links ]
43 Tao H, Wenjun L, Brandi N, Simmons H, Harris T and Massiah M. 2010. Purifying natively folded proteins from inclusion bodies using sarkosyl, triton X-100, and CHAPS. Benchmarks. 48: 61 - 64. [ Links ]
44 Tsumoto K, Abe R, Ejima D and Arakawa T. 2010. Non - denaturing solubilization of inclusion bodies. Current Pharmaceutical Biotechnology. 4: 309 - 312. [ Links ]
45 Wall G and Pluckthun A. 1995. Effects of overexpressing folding modulators on the in vivo folding of heterologous proteins in Escherichia coli. Current Opinion in Biotechnology. 6: 507-516. [ Links ]
46 Wood t and Peretti s. 1991. Effect of chemically-induced, cloned-gene expression on protein synthesis in E. coli. Biotechnology and Bioengineering. 38: 397-412. [ Links ]
47 Yoder J and Dormitzer P. 2006. Alternative intermolecular contacts underlie the rotavirus VP5* two - to - three fold rearrangement. The EMBO Journal. 25: 1559 - 1568. [ Links ]
48 Zárate S, Espinosa R, Romero P, Méndez E, Arias C and López S. 2000. The VP5 domain of VP4 can mediate attachment of rotaviruses to cells. Journal of Virology. 74: 593 - 599. [ Links ]
