










Services on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Biotecnología
Print version ISSN 0123-3475
Rev. colomb. biotecnol vol.17 no.2 Bogotá July/Dec. 2015 Epub Aug 10, 2021
https://doi.org/10.15446/rev.colomb.b¡ote.v17n2.48522
Artículos cortos
Estandarización de un protocolo para Northern blot no radioactivo usado en la detección de pequeños RNA en células Vero
* Grupo de Medicina Molecular y de Translación. Faculta de Medicina. Universidad de Antioquia, Colombia. naticampillo@gmail.com, jp.salazar04@gmail.com, juanc.gallegomez@gmail.com.
El interés en la detección, identificación, y caracterización funcional de los pequeños RNAs no codificantes (sRNAs), ha generado la necesidad de optimizar las metodologías comúnmente usadas en su detección, la reacción en cadena de la polimerasa cuantitativa (RT-qPCR) y Northern blot, con el fin de que sean más sensibles y específicas. A pesar de la baja sensibilidad del Northern blot, esta metodología continúa siendo de uso común en la detección de sRNAs porque permite detectar el RNA pequeño así como a sus precursores, razón por la cual se usa como una metodología complementaria en este tipo de investigaciones. En este trabajo se describe la implementación de un nuevo protocolo para Northern blot no radioactivo, con modificaciones dirigidas a mejorar su sensibilidad y especificidad. El diseños de la sonda con la tecnología LNA, el marcaje de esta con Digoxigenina y por último la fijación del RNA a la membrana mediante 1-Ethyl-3-(-3-dimethyla-minopropyl) carboniimide (EDC) y finalmente se discuten los fundamentos teóricos de estos cambios.
Palabras clave: 1-ethyl-3-(-3-dimethylaminopropyl) carboniimide); Digoxigenina; Northern blot no radioactivo; Sondas LNA; sRNAs
The interest in detection, identification and functional characterization of small non-coding RNAs (snRNAs), has generated the need to optimize the methodologies commonly used in its detection in specificity and sensitivity, The Quantitative reverse transcription PCR (RT-qPCR) and Northern blot. Even though the low sensitivity of Northern blot, this method continues to be commonly used in the sRNAs, because its capacity to detect the sRNA and its precursor, which is the reason why Northern blot is used as complementary method in this sort of Research.
This work describes the implementation of an innovative non-radioactive Northern blot protocol, with modifications that improving the sensibility and specificity, with the discussion of the theoretical foundations of such modifications.
Key words: 1-ethyl-3-(-3-dimethylaminopropyl) carboniimide); Digoxigenine; Northern blot non-radioactive; LNA probe; sRNAs
Introducción
Los pequeños RNA no codificantes (sRNAs, small non-coding RNAs) son secuencias involucradas en diversas funciones celulares, tanto constitutivas como de regulación. Desde el descubrimiento del primer RNA pequeño, lin-4 (Lee et al., 1993; Wightman et al., 1993), la lista de pequeños RNAs no codificantes ha aumentado, reportándose la participación de alguno de ellos en procesos biológicos como corte y empalme de exones, traducción genética, desarrollo, diferenciación, muerte celular, control metabólico, defensa antiviral, regulación de expresión genética transcripcional y post-transcripcional (Gomes et al., 2013; van Wolfswinkel & Ketting, 2010; Winter et al., 2009). La función para muchos de estos sRNAs aún se desconoce, por lo cual el interés y la necesidad de su detección, caracterización y validación funcional continúan.
Las metodologías utilizadas para la detección de los sRNAs, enfrentan grandes desafíos impuestos por su tamaño (≤200 nucleótidos) y el bajo nivel de expresión de los pequeños RNAs, que en algunos casos no supera el umbral de detección de las técnicas usadas hasta el momento. Por las limitaciones anteriormente mencionadas, los métodos más utilizados para la detección de sRNAs (RT-qPCR y Northern blot) deben ser constantemente optimizados para mejorar la sensibilidad, especificidad de detección y cuantificación.
El Northern blot es una metodología directa que permite la detección de los pequeños RNAs, sus secuencias precursoras o intermediarias, evaluar sus propiedades de expresión y determinar su tamaño Wang & Yang (2010) razón por la cual sigue siendo ampliamente usado. El proceso general del Northern blot consiste en extraer RNA total, separarlo por tamaño mediante electroforesis y transferir e inmovilizar en una superficie sólida (membrana). La sonda con secuencia complementaria al RNA de interés, es marcada con métodos radioactivos o no radioactivos, luego se incorpora en la membrana que contiene el RNA inmovilizado y finalmente la hibridación con la secuencia complementaria es detectada.
Para la metodología Northern blot se han publicado diferentes protocolos que varían principalmente en el diseño y marcaje de la sonda, donde comúnmente se utilizan sondas de DNA marcadas con fósforo radioactivo (32P) (Gurman S Pall & Hamilton, 2008; Várallyay et al., 2008). Un nuevo protocolo para Northern blot no radiactivo denominado LED (LNA-EDC-DIG), fue reportado Kim et al. (2010), integra tres metodologías desarrolladas separadamente, que modifican a su vez tres aspectos del proceso determinantes de la especificidad y sensibilidad del Northern blot. Las modificaciones consisten básicamente en remplazar las sondas de oligonucleótidos de DNA, por sondas que contienen ácidos nucleicos bloqueados o no accesibles (Locked Nucleic Acid, LNA). La inmovilización del RNA en la membrana, que normalmente se hace con luz ultravioleta (UV), es remplazado por el uso de EDC (1-Ethyl-3-(-3-dimethylaminopropyl) carboniimide)). Adicionalmente, el uso de radioisótopos (32P), el método más común de marcaje de sondas, con el cual se deben asumir todos los riegos de la radioactividad, es sustituido por el uso de Digoxigenina (DG).
Este nuevo protocolo aumenta la sensibilidad y especificidad en la detección de pequeños RNAs por Northern blot, por lo que su uso e implementación en los estudios de detección, caracterización y funcionalidad de nuevos sRNAs será de gran utilidad. Por lo anterior, la descripción de la implementación y modificaciones realizadas de este protocolo usado en la detección de un RNA pequeño, así como la explicación y discusión de los fundamentos teóricos de las modificaciones planteadas son el objetivo de este trabajo.
Materiales y Métodos
Extracción de RNA total a partir de células Vero
Todas las extracciones de RNA total se hicieron con TRI-reagent (Sigma™), de manera breve 4x106 células Vero fueron lavadas con PBS 1X para eliminar proteínas, se re-suspendieron en 1ml de Trizol, se incubaron 3 minutos (min) a temperatura ambiente (TA), se adicionó 200|jl de Cloroformo (Merck™) y se centrifugó a 5000 revoluciones por minuto (rpm) durante 15 min, se tomó la fase acuosa y se adicionó Isopropanol frío, se incubó durante 10 min a -20°C, nuevamente se centrifugó a 5000 rpm durante 10 min usando microcentrífuga (Eppendorf™, 5810 R). El pellet se lavó con 1ml de Etanol al 75% con vortex suave, se centrifugó nuevamente a 10.700 rpm durante 5 min, se dejó secar el etanol a TA y posteriormente se re-suspendió el pellet en 100|l de agua tratada con DEPC. Con NanoDrop 2000 (Thermo Scientific™) se determinó pureza y concentración del RNA extraído.
Northern blot no radioactivo
El RNA extraído, fue evaluado por Northern blot no radioactivo. Para esto se modificó, implementó y estandarizó el protocolo para detectar miRNAs, previamente descrito Kim et al. (2010), el protocolo se desarrolló en 4 pasos (figura 1): 1) Separación del RNA en gel de poliacrilamida, 2) Transferencia e inmovilización del RNA, 3) Pre-hibridación e Hibridación de la sonda, 4) Detección del RNA. Para implementar el protocolo se usó una sonda específica y complementaria al RNA pequeño nuclear U6 (snRNA U6, del inglés small nuclear RNA) conteniendo nucleótidos modificados LNA (Exiqon™), como control negativo se usó una sonda también de nucleótidos modificados LNA (Exiqon), pero en una secuencia desorganizada "Scramble".
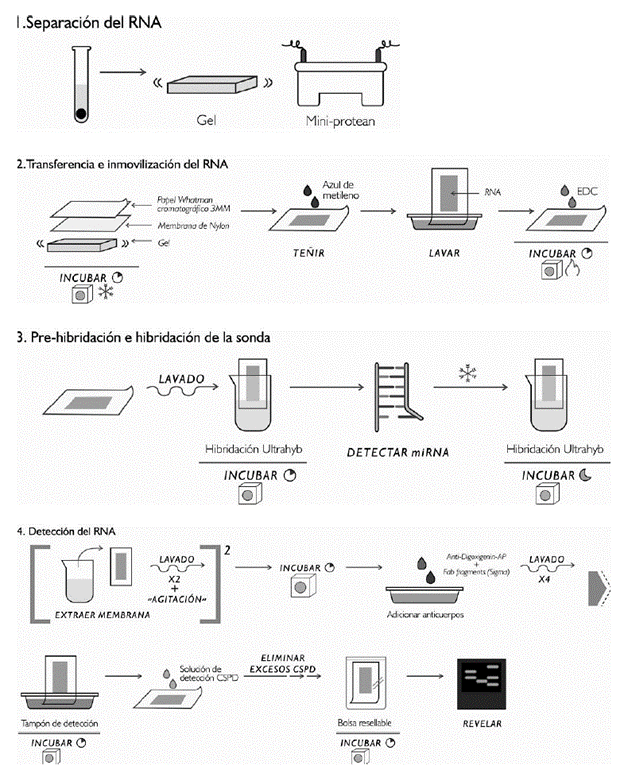
Separación del RNA. Se hizo un corrido electro-forético en gel de poliacrilamida 15% (desnaturalizante 7M de úrea, Sigma™) utilizando el sistema Mini-protean (BioRad™, Hercules, CA, USA) y el marcador de peso molecular para miRNAs (New England BioLabs™ - miRNA marker). 2μg de RNA extraído, se mezclaron con tampón de carga (Gel Loading Buffer II, Sigma™), las muestras y 5μl de marcador de peso molecular, se desnaturalizaron a 95°C durante 1 min, luego se cargaron en el gel y se corrieron aplicando 12.5 v/cm, en cámara de electroforesis MiniProtean (Biorad™).
Transferencia e inmovilización del RNA. Una vez finalizado el corrido electroforético, el RNA separado fue transferido a una membrana de Nyloncargada positivamente (Roche™), utilizando el equipo Trans-Blot SD Semi-Dry Transfer Cell (Bio-Rad™), en este equipo se hizo un montaje tipo sándwich con 3 hojas de papel Whatman cromato-gráfico 3MM (Whatman), la membrana de Nylon, el gel y finalmente 3 hojas más de papel cromato-gráfico, luego se adicionó TBE 1X (89mM Tris base, 89mM ácido borico, 2mM EDTA). Para garantizar la transferencia, esta se realizó en cuarto frío a 15V durante 60 min. Transcurridos el tiempo de la transferencia, se evaluó si hubo transferencia del RNA a la membrana, tiñéndola con azul de metileno (0.02% azul de metileno, 0.3M NaOAc pH 5.5) durante 3 a 10 min, en este tiempo se observaron las bandas de RNA en la membrana, luego el azul de metileno se lavó con agua destilada. Posteriormente se inmovilizó el RNA en la membrana, para lo cual se humedeció una hoja de papel cromato-gráfico Whatman con solución EDC (para 24ml de EDC: 0.753gr de 1-ethyl-3-(3-dimethylaminopropyl) carboniimide - Sigma, 245 |i de 1 Methylmidazole 12.5M, se aforó al volumen final en agua tratada con DEPC), y se incubó durante 1 hora a 65°C.
Pre-hibridación e hibridación de la sonda. Transcurrida la hora de inmovilización la membrana se lavó con Agua desionizada. Se precalentó a 65°C el tampón de hibridación Ultrahyb (Ambion), a cada botella de hibridación se adicionó 15ml del tampón, se introdujo la membrana conteniendo los RNA inmovilizados y se incubó durante 30 min a 37°C para pre-hibridar. Las sondas marcadas con Digoxigenina y Estreptavidina, para detectar el miRNA y el marcador de peso molecular, respectivamente, se desnaturalizaron a 95°C durante 1 min, se pusieron en hielo inmediatamente, se adicionaron a la botella de hibridación conteniendo la membrana con el RNA y se incubaron nuevamente a 37°C durante toda la noche.
Detección del RNA. Para realizar el proceso de detección se retiró la membrana de la botella de hibridación, se lavó dos veces con tampón de baja astringencia (2x SSC con 0.1% (p/v) SDS) a 37°C con agitación durante 15 min. Dos veces con tam-pón de alta astringencia (0.1x SSC con 0.1% (p/v) SDS) a 37°C durante 5 min. Una vez con tampón de lavado (1x SSC) durante 10 min, luego se incubó a 37°C durante 3h en tampón de bloqueo (DIG wash and Block buffer set, Roche). Posteriormente se adicionaron los anticuerpos, Anti-Digoxigenin-AP, Fab fragments (Sigma) en dilución 1:15.000 y Estreptavidina (Promega) 1:4000, se incubaron durante 30min a TA. Después de incubar los anticuerpos la membrana se lavó con tampón de lavado (DIG wash and Block buffer set, Roche) cuatro veces durante 15 min a TA. Se incubó la membrana 5 min en Buffer de detección (DIG wash and Block buffer set, Roche) a TA, luego sobre el lado de la membrana que contiene el RNA, se adicionó la solución de detección CSPD (Roche) en dilución 1:100 se dejó a TA durante 5 min, posteriormente se eliminó el exceso de CSPD de la membrana y se introdujo en una bolsa sellable y resistente al calor (heat - seleable bag) se incubó durante 15 min a 37°C, para proceder a su detección con cassette de revelado y películas de alta sensibilidad. Las películas se expusieron durante 30min y 1h para luego ser reveladas.
Resultados y discusión
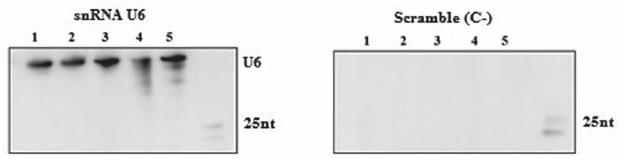
Con el protocolo descrito anteriormente se observó señal cuando el RNA fue evaluado con la sonda específica del snRNA U6 (figura 2), un RNA pequeño nuclear involucrado en el ensamblaje y funcionamiento del complejo de corte y empalme celular, con un tamaño aproximado de 150 nucleótidos (nt). Cuando se evaluó el mismo RNA con la sonda de control negativo Scramble (con nucleótidos desorganizados) no se observó señal (figura 5), lo que indica que la señal observada con la sonda complementaria al snRNA U6 es específica. Con lo anterior se demuestra la sensibilidad y especificidad del protocolo en la detección de un snRNA constitutivo celular.
Como se mencionó inicialmente en este protocolo se proponen cambios en tres aspectos que contribuyen a la especificidad, sensibilidad y seguridad de esta técnica, como son el tipo de sonda, su marcaje y el proceso de inmovilización del RNA en la membrana. Para entender la razón de las modificaciones realizadas y con el fin de que este protocolo pueda ser implementado y modificado posteriormente, a continuación se explicarán y discutirán los fundamentos teóricos de los cambios realizados en el mismo.
Diseño de sondas
Cuando se diseñan y sintetizan moléculas similares a los ácidos nucleicos, como las sondas utilizadas en Northern blotting, tres características generales deben tenerse en cuenta: primero, las moléculas diseñadas deben ser eficientes en la oligomerización. Segundo, deben tener alta afinidad y reconocimiento selectivo del ácido nucleico complementario; esta propiedad puede ser medida por los valores de temperatura de fusión (Tm), con altos valores menor afinidad, valores bajos mayor afinidad. Tercero, tener buena solubilidad en agua (Wengel 1999).
Para obtener sondas con oligomerización eficiente y alta afinidad en la hibridación con las cadenas complementarias de los ácidos nucleicos, estas deben ser diseñas de forma tal que la estructura de cada una de estas sondas tenga una configuración estructural lo más similar posible a la encontrada en los ácidos nucleicos naturalmente Petersen et al. (2002). Los ácidos nucleicos naturalmente tienen dos configuraciones estructurales, el tipo A (también llamado tipo N) y el tipo B (también llamado tipo S). Estas conformaciones están dadas por los cambios de estructura en los azúcares de los nucleótidos (Wenge 1999). La ribosa (azúcar del RNA) normalmente se encuentra con configuración del tipo A, mientras que el DNA puede encontrarse en cualquiera de las dos configuraciones dependiendo del ambiente (fuerza iónica y humedad relativa) (Wengel 1999) (figura 3).
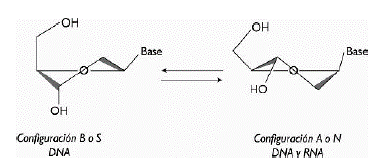
Figura 3 Esquema mostrando las configuraciones estructurales de los ácidos nucleicos. Configuración B o S, en al que se encuentra el DNA. Configuración A o N en la que se encuentran tanto el DNA como el RNA. Imagen tomada y modificada de Wengel, 2009.
Por su parte los LNA (Locked Nucleic Acids) son nucleótidos modificados, su estructura se caracteriza por tener un puente de metileno que conecta el 2'-Oxi-geno con el 4'-Carbono de la ribosa (figura 4). Este puente resulta en una conformación bloqueada C3'-endo, impidiendo que el nucleótido adopte otra configuración diferente a la A, configuración en la que se encuentra el RNA naturalmente (Gurman S Pall & Hamilton, 2008; Wengel 2009).
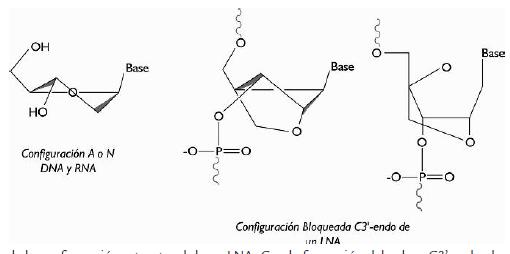
Figura 4 Ilustración de la configuración estructural de un LNA. Con la formación del enlace C3'-endo el nucleótido permanece con configuración A o N, forma estructural en la que se encuentra el RNA naturalmente. Imagen tomada y modificada de (Wengel, 2009).
Esto hace que las sondas LNA usadas en el protocolo propuesto por Kim et al., 2010 tengan alta afinidad y estabilidad térmica representada en bajos valores de Tm y mayor especificidad.
Inmovilización del RNA en la membrana
La inmovilización del RNA en la membrana inducida por UV sucede por el entrecruzamiento entre fracciones de residuos de uracilo y los grupos amino cargados positivamente presentes en la membrana Sambrook et al. (2001). El proceso tiene tres desventajas que afectan notablemente la detección de pequeños RNAs. Ocurre degradación por UV de los grupos funcionales (grupos amino de la membrana) requeridos para el apareamiento de bases. Los RNA se fragmentan, disminuyendo su longitud impidiendo conservar las secuencias completas, limitándose así la hibridación de las sondas. Como última desventaja, tenemos que el número de bases involucradas no puede ser controlado en la reacción inducida por UV, por esto podría ocurrir un entrecruzamiento excesivo, reduciendo la disponibilidad de bases para la hibridación con una sonda complementaria.
A pesar de lo anterior el entrecruzamiento entre RNA y membrana inducido por UV es rápido, poco costoso y eficiente para RNA con más de 70nt Gurman S Pall & Hamilton (2008). Sin embargo lo ideal para la detección por hibridación de pequeños RNAs, debe ser el acoplamiento de la secuencia de forma tal que permanezca completamente disponible para la hibridación con la sonda complementaria (Gurman et al., 2008; Gurman et al., 2007). El entrecruzamiento por medio del extremo 5' (diferentes modificaciones pueden encontrarse en el extremo 3', por esto se prefiere el 5' para un entrecruzamiento) de los pequeños RNAs, y no a partir de las bases nitrogenadas como sucede usando UV, es una alternativa. Este tipo de en-trecruzamiento es posible realizarlo usando 1- Ethyl-3 (3-dimetilaminopropil) Carboniimida (EDC), compuesto que previamente ha sido usado para entrecruzar el extremo 5' de oligonucleótidos sintéticos en diferentes substratos sólidos Gurman Singh Pall et al. (2007). El uso del EDC en la inmovilización del RNA en la membrana dirigido a la detección de miRNA surge como una alternativa que mejora de manera notable la sensibilidad en la detección de estas pequeñas moléculas.
Marcaje de sondas
El análisis de transferencia de Northern blot usando radioisótopos es la metodología usada actualmente para la detección de sRNAs, sin embargo, esta técnica generalmente es dispendiosa, costosa, implica riesgos ambientales y laborales, adicionalmente en nuestro país existe una justificada y seria exigencia para no promover el uso de radioisótopos. Como una alternativa, el sistema de marcaje (DIG)-Digoxigenina basa su sistema de detección en el proceso de quimioluminiscencia, lo que genera varias ventajas en comparación a las técnicas de marcado radioactivo, como: alta sensibilidad, tiempos cortos de exposición, vida útil más larga, y mayor seguridad (Ramkissoon et al., 2006).
Conclusión
La creciente necesidad e interés de identificar, detectar, caracterizar y validar nuevos sRNAs, ha ido de la mano con la optimización y mejora de las metodologías utilizadas para tales fines, dándose lugar a protocolos como el descrito previamente. El conocer y entender los fundamentos teóricos por los cuales se optimizan protocolos como el descrito en este trabajo, aporta al desarrollo, planteamiento de nuevas optimizaciones, a la creación de nuevos protocolos y metodologías que puedan ser consideradas en el desarrollo de investigaciones de este tipo.
Agradecimientos
Los autores agradecen al Departamento Administrativo de Ciencia Tecnología e Innovación -COLCIENCIAS- por su apoyo en la realización de este trabajo, mediante la financiación del proyecto 111554531621. NCP tuvo financiación también de Colciencias como Joven Investigadora, durante el período 2010-2011. JCGG agradece a la Universidad de Antioquia, por la concesión de su Dedicación Exclusiva 2014-2015 para la Facultad de Medicina.
REFERENCIAS
Gomes, A. Q., Nolasco, S., & Soares, H. (2013). Non-coding RNAs: multi-tasking molecules in the cell. International Journal of Molecular Sciences, 14(8), 16010-39. [ Links ]
Kim, S. W., Li, Z., Moore, P. S., Monaghan, a P., Chang, Y., Nichols, M., & John, B. (2010). A sensitive non-radioactive northern blot method to detect small RNAs. Nucleic Acids Research, 38(7), e98. [ Links ]
Lee, R. C., Feinbaum, R. L., & Ambros, V. (1993). The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell, 75, 843-854. [ Links ]
Pall, G. S., Codony-Servat, C., Byrne, J., Ritchie, L., & Hamilton, A. (2007). Carbodiimide-mediated cross-linking of RNA to nylon membranes improves the detection of siRNA, miRNA and piR-NA by northern blot. Nucleic Acids Research, 35(8), e60. [ Links ]
Pall, G. S., & Hamilton, A. J. (2008). Improved northern blot method for enhanced detection of small RNA. Nature Protocols, 3, 1077-1084. [ Links ]
Petersen, M., Bondensgaard, K., Wengel, J., & Peter Jacobsen, J. (2002). Locked nucleic acid (LNA) recognition of RNA: NMR solution structures of LNA: RNA hybrids. Journal of the American Chemical Society, 124, 5974-5982. [ Links ]
Ramkissoon, S. H., Mainwaring, L. A., Sloand, E. M., Young, N. S., & Kajigaya, S. (2006). Nonisotopic detection of microRNA using digoxigenin labeled RNA probes. Molecular and Cellular Probes, 20, 1-4. [ Links ]
Sambrook, J., Fritsch, E. F., & Maniatis, T. (2001). Molecular Cloning: A Laboratory Manual. Vol. 3. Cold Spring Harbor Laboratory. [ Links ]
Van Wolfswinkel, J. C., & Ketting, R. F. (2010). The role of small non- coding RNAs in genome stability and chromatin organization. Journal of Cell Science, 123(Pt 11), 1825-39. [ Links ]
Várallyay, E., Burgyán, J., & Havelda, Z. (2008). MicroRNA detection by northern blotting using locked nucleic acid probes. Nature Protocols, 3(2), 190-6. [ Links ]
Wang, Z., & Yang, B. (2010). MicroRNA Expression detection methods. Heidelberg: Springer-Verlag. [ Links ]
Wengel, J. (1999). Synthesis of 3'-C-and 4'-C-Branched Oligode-oxynucleotides and the Development of Locked Nucleic Acid (LNA). Acc. Chem. Res ., 32, 301 -310. [ Links ]
Wengel, J. (2009). Locked Nucleic Acid Technology : A brief overview. Exiqon, 7. [ Links ]
Wightman, B., Ha, I., & Ruvkun, G. (1993). Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell, 75, 855-862. [ Links ]
Winter, J., Jung, S., Keller, S., Gregory, R. I., & Diederichs, S. (2009). Many roads to maturity: microRNA biogenesis pathways and their regulation. Nature Cell Biology, 11(3), 228-34. [ Links ]
Recibido: 21 de Enero de 2015; Aprobado: 23 de Octubre de 2015
 This is an open-access article distributed under the terms of the Creative Commons Attribution License
This is an open-access article distributed under the terms of the Creative Commons Attribution License
