Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
PermalinkINTRODUCCIÓN
Los agentes bloqueadores beta-adrenérgicos son amino -alcoholes quirales usados en el tratamiento de la hipertensión arterial y se comercializan como mezcla racémica. Uno de los enantiómeros posee el efecto terapéutico deseado (generalmente el enantiómero S), mientras que el otro suele ser menos activo o inducir efectos secundarios (e.g. broncoconstricción) (S Bangalore, 2000; Sripal Bangalore, Messerli, Kostis, & Pepine, 2007; Sripal Bangalore, Parkar, Grossman, & Messerli, 2007). El propranolol ((R/S)-1-(isopropilamino)-3-(naftalen-l-iloxi)propan -2-ol) es un ejemplo, por esta razón el diseño de estrategias para separar mezclas racémicas es importante.
Las lipasas, triacilglicerol éster hidrolasas, E.C.3.1.1, catalizan la hidrólisis de triglicéridos en las células. También pueden catalizar reacciones de transferencia de grupos acilo, formación de perácidos y aminólisis, entre otras, siendo unas de las enzimas más utilizadas en la industria (Ansorge-Schumacher & Thum, 2013; Naik et al., 2010; Turki, 2013). Las reacciones catalizadas por lipasas ofrecen una vía amigable con el medio ambiente para la síntesis de compuestos enantiopuros (Ghanem, 2007). El éxito de estas reacciones depende de la habilidad de la enzima para discriminar entre los dos enantiómeros de una mezcla racémica (Ghanem & Aboul-Enein, 2004).
Una de estas lipasas, la lipasa B de Candida antarctica (CalB) se ha utilizado en la resolución cinética de múltiples sustratos, como: ésteres de profeno (Qin et al., 2013), alcoholes secundarios (Ursoiu, Paul, Kurtán, & Péter, 2012), aminas (Busto, Gotor-Fernández, & Gotor, 2011) y en la acilación quimioselectiva de aminoácidos (Ferrari et al., 2014), aminoalcoholes (Le Joubioux et al., 2013) y flavonoides (Mellou, Loutrari, Stamatis, Roussos, & Kolisis, 2006). CalB tiene enantioselectividad moderada (£=63) por el R-propranolol y quimioselectividad por su grupo OH, cuando se utiliza acetato de vinilo como donador acilo y tolueno como solvente (Escorcia, Molina, Daza, & Doerr, 2013). Sin embargo, este valor de enantioselectividad es mayor que el de las lipasas de Pseudomonas cepacia, Rhizopus niveus y Pseudomonas fluorescens (Barbosa, Ariza, Ortiz, & Torres, 2010).
CalB es una proteína globular de 317 residuos con plegamiento alfa/beta (9 láminas beta rodeadas por 10 hélices alfa). La entrada de su sitio activo está definida por tres hélices alfa. CalB presenta la tríada catalítica aspartato (D187), histidina (H224) y serina (S105) (Uppenberg et al., 1995; Uppenberg, Hansen, Patkar, & Jones, 1994) y un mecanismo catalítico similar al de las proteasas de serina (Ghanem, 2007). La H224 se encuentra localizada de tal manera que su cadena lateral queda expuesta en el sitio activo justo en medio de D187 y S105. El entorno de S105 es de naturaleza polar; además de H224 se encuentran los residuos T40, Q106, D134 y Q157 (figura 1). Los residuos T40 y Q106 forman el hueco oxianiónico (figura 2).
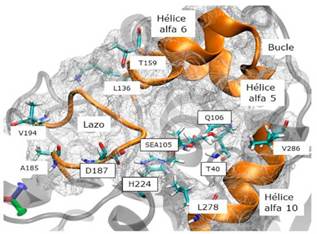
Figura 1 Estructuras secundarias que rodean al sitio activo de AcetilCalB. i) Residuos aminoacídicos L136 a T159, corresponden a la hélice alfa 5, bucle y hélice alfa 6. ii) Lazo compuesto por los residuos aminoacídicos A185 a V194. iii) Segmento de la hélice alfa 10, compuesta por los residuos aminoacídicos L278 a V286. Los residuos aminoacídicos que componen la triada catalítica (D187, H224 y SEA105), y los residuos aminoacídicos que componen el hueco oxianiónico (T40 y Q106) se indican con un recuadro. Esta representación corresponde a la AcetilCalB en el complejo de Michaelis OS1-I.
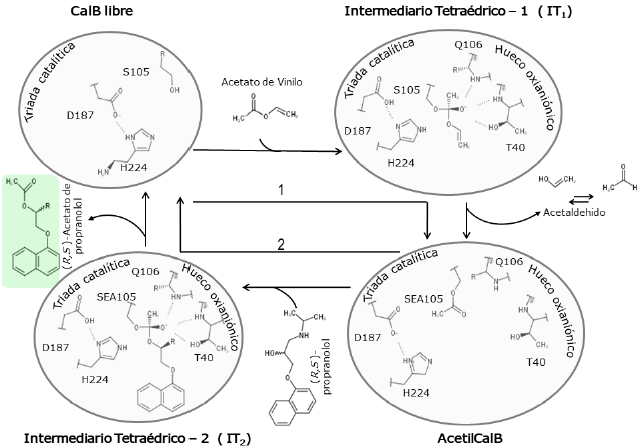
Figura 2 Modelo del mecanismo de acilación de propranolol catalizado por CalB indicando los dos intermediarios tetraédricos y la AcetilCalB. 1 Reacción de acilación de la enzima. 2 Reacción de desacilación de la enzima. El primer sustrato, donador acilo, es el acetato de vinilo. El propranolol es el segundo sustrato. El producto final de la reacción es el (R,5)-acetato de propranolol.
El modelo del mecanismo catalítico de la acilación del propranolol catalizado por CalB incluye la acilación y desacilación del residuo S105 e involucra dos intermediarios tetraédricos (IT1 e IT2, figura 2). El primero es el resultado del ataque nucleofílico de la serina catalítica al sustrato acilante y el producto es el complejo enzima acilada (AcetilCalB). El segundo intermediario tetraédrico es el producto del ataque nucleofílico del -OH del propranolol (sustrato aceptor del acilo) a la acil-enzima que conlleva finalmente a la formación del éster del propranolol y a la restitución de la enzima (Escorcia, 201 5; Escorcia et al., 2013). La enantioselectividad de CalB se origina en la reacción de transferencia del grupo acilo de SEA105 al propranolol (Escorcia, 2015; Escorcia, Daza, & Doerr, 2014). Los intermediarios tetraédricos son considerados análogos estructurales y energéticos de los estados de transición en las reacciones catalizadas por CalB (Bocola, Otte, Jaeger, Reetz, & Thiel, 2004; García-Urdiales, Ríos-Lombardía, Mangas-Sánchez, Gotor-Fernández, & Gotor, 2009; Nyhlén, Martín-Matute, Sandstróm, Bocola, & Backvall, 2008; Xu et al., 2010). Previo a la formación de los intermediarios tetraédricos se forman complejos enzima-sustrato estables denominados complejos de Michaelis (MCC, figura 3).
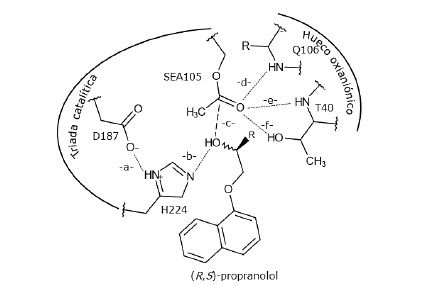
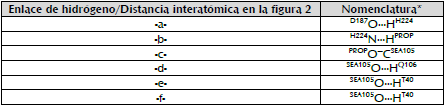
Figura 3 Modelo de los complejos de Michaelis CalB-propranolol en el paso enantioselectivo de la reacción de acilación (Escorcia et al., 2014; Escorcia, Sen, Daza, Doerr, & Thiel, 2017). Las líneas punteadas indican los enlaces de hidrógeno claves en el mecanismo catalítico. La línea discontinua indica distancia entre el oxígeno del grupo hidroxilo del propranolol y el carbono electrodeficiente de SEA105 (-c-). Todas las longitudes de enlace indicadas deben ser menores o iguales a 4,0 A. (ver texto).
De acuerdo con nuestros modelos previamente reportados (Escorcia et al., 2014, 2017), estos complejos son estabilizados por enlaces de hidrógeno entre el propranolol y los residuos de la triada catalítica y del hueco oxianiónico, -a-, -b-, -d-, -e- y -f- (figura 3). Los enlaces de hidrógeno -d-, -e- y -f- permiten evaluar la estabilización del oxígeno carbonílico de la S105 acilada (SEA105), (líneas punteadas, figura 3). La cercanía del grupo OH del propranolol (PROP) a la triada catalítica se evalúa por la distancia -c- (línea discontinua, figura 3). Los ángulos asociados a estos enlaces de hidrógeno son: ángulo A (D187O...H224H-H224N), ángulo B (H224N-PROPH...PROPO), ángulo C (PR°PO-SEA105C-SEA105O), ángulo D (sEA105o...Q106H-Q106N), ángulo E (SEA105°...T40H-T40O), y ángulo F (SEA105°...T40H-T40N). Un complejo AcetilCalB-propranolol se considera un MCC cuando cumple con los siguientes criterios: i) Enlace de hidrógeno -b-, menor o igual a 4,0 A. ii) Longitud del enlace -c- menor o igual a 4,0 A. iii) Todos los enlaces de hidrógeno -a -, -d-, -e- y -f- deben tener valores menores o iguales a 4,0 A (Escorcia et al., 2014, 201 3; Ferrari et al., 201 4) (figura 3).
Un análisis de los MCC y de los ITs en el paso enantioselectivo de la acilación del (R,S)-propranolol mediante la combinación de acoplamiento molecular y Dinámica Molecular (DM) evidenció que el propranolol puede orientarse en el sitio activo de CalB de dos maneras: modo I y modo II (Escorcia et al., 2014) (figura 4).
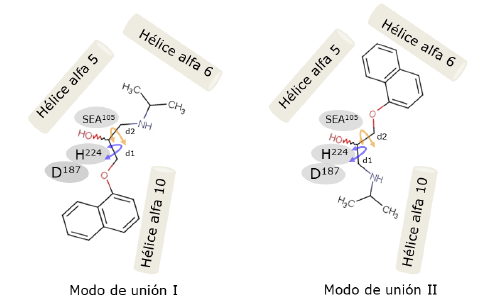
Figura 4 Modos de unión del (R,5)-propranolol en el sitio activo de AcetilCalB. Los residuos de la triada catalítica (D, H y SEA) se ubican en el medio de las hélices alfa que rodean al sitio activo (Escorcia et al., 2014). Los ángulos diedros d1 (OCCO) y d2 (OCCN) dan flexibilidad para que el propranolol se ubique en los dos modos de unión y son representados por flechas.
Lo anterior se debe a que el grupo OH en el (R,S)-propranolol se encuentra en la parte central del eje más largo de la molécula y a que ángulos diedros di OCCO y d2 OCCN dan flexibilidad para que el propranolol se ubique en los dos modos de unión (Escorcia et al., 2014) (figura 4).
Los MCC que tienen una conformación similar a la del estado de transición se denominan complejos de ataque cercano (NACs) (Bruice & Benkovic, 2000; Lightstone & Bruice, 1996). En estos complejos, los átomos que intervienen directamente en la formación o rompimiento del enlace en la reacción de acilación y desacilación deben tener una longitud de enlace menor o igual a la sumatoria de sus radios de van der Waals y un ángulo de ataque al grupo carbonilo en el rango de +/- 20° alrededor de un eje imaginario ubicado a ~107° del plano del grupo carbonilo (Bruice, 2002, 2006) (figura 5). El análisis de los NACs ha permitido en varios casos explicar el origen de la catálisis o reproducir el efecto catalítico (Giraldo, Roche, Rovira, & Serra, 2006; Griffin et al., 2012; Mazumder-Shivakumar, Kahn, & Bruice, 2004; Strajbl, Shurki, Kato, & Warshel, 2003); específicamente en CalB, este enfoque se ha usado para predecir la capacidad de catalizar reacciones tipo Diels-Alder (Linder, Hermansson, Liebeschuetz, & Brinck, 2011). El análisis de las poblaciones de NACs en la reacción de acilación del (R,S)-propranolol permite dar una explicación plausible a la enantioselectividad experimental. La población de NACs encontrada en varias conformaciones del S-propranolol fue ligeramente menor que la población de NACs encontrada en los confórmeros de R-propranolol (Escorcia et al., 2014). Esta explicación se basó en la suposición de que a mayor población de NACs mayor velocidad de la reacción enzimática (Lightstone & Bruice, 1996).
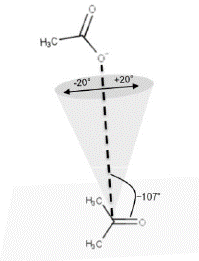
Figura 5 Conformación de ataque cercano (NACs). La distancia (línea discontinua en negrita) entre el oxígeno con carga formal -1 y el carbono electrodeficiente (átomos que harán parte en la reacción) se encuentran a una distancia menor o igual que la sumatoria de sus radios de van der Waals y con un ángulo de ataque al grupo carboni-lo, de +/- 20° alrededor de un eje imaginario que está a ~107° del plano del grupo carboxilo (Bruice, 2002, 2006).
Con miras a ampliar nuestra comprensión sobre las poblaciones de NACs encontramos que pueden variar hasta en un 50% cuando se cambia la distribución de velocidades iniciales asignadas a los átomos durante la DM (Torres & Bruice, 2000). Además, que los cambios en las velocidades iniciales originan diferencias notorias entre réplicas de trayectorias inclusive usando el mismo campo de fuerza y estructura de partida (Cino, Choy, & Karttunen, 2012). Para profundizar en la comprensión de la función de los NACs en la enantioselectividad de la acilación del (R,S)-propranolol catalizada por CalB realizamos un estudio más detallado de los MCCs y de los NACs que el publicado previamente (Escorcia et al., 2014). Aumentamos el muestreo conformacional incrementando el tiempo de simulación de la DM QM/MM en un 66% (de 1,5 a 2,5 ns), y el número de simulaciones realizadas con diferentes valores de siembra del generador de números aleatorios del 50% (de tres a seis). Nuestros resultados muestran que la evolución de los MCC, y el tiempo de vida de los NACs depende estrechamente del muestreo conformacional realizado.
MÉTODOS
Se realizó un estudio detallado de los MCCs y de los NACs del paso enantioselectivo de la reacción de acilación del (R,S)-propranolol utilizando 6 distribuciones de velocidades iniciales y simulaciones de 2,5 ns, con un paso de 1 fs, y temperatura constante, 300 K, para la fase productiva. Se analizó detalladamente la evolución de los enlaces de hidrógeno claves en el mecanismo catalítico, las poblaciones de NACs, y la distancia entre el oxígeno del grupo hidroxilo del propranolol y el carbono electrodeficiente de SEA105.
-Selección de las distribuciones de las velocidades iniciales de los átomos en la Dinámica Molecular El generador de números aleatorios se usa en las simulaciones de DM para asignar las velocidades iniciales en los átomos. Este generador necesita de un valor entero (valor de siembra) para iniciar la producción de números aleatorios. Cada valor de siembra permite generar una secuencia de números aleatorios única. En el programa CHARMM el valor de la siembra (parámetro ISEED number) puede ser modificado. Se utilizaron los números de siembra (random seed): 55627, 1238, 384555, 835, 234 y se incluyó el valor por omisión en CHARMM que es 314159. Los números 835, 234 y 314159 fueron, también, empleados en un estudio previo (Escorcia et al., 2014).
-Selección de los Complejos de Michaelis (MCCs)
Las estructuras iniciales de los MCC fueron seleccionadas de un trabajo previamente reportado por nuestro grupo (Escorcia et al., 2013). En el modo de unión I se seleccionaron dos MCCs para el enantiómero R y tres para el enantiómero S y se les asignaron las etiquetas OR1-I, OR2-I y OS1-I, OS2-I, OS3-I. En el modo de unión II se eligió el único MCC reportado para cada uno de los enantiómeros y se etiquetaron como OR3-II y OS4-II (figura A1, información adicional). A continuación, se explica brevemente la metodología empleada en el modelado de las estructuras iniciales de los MCC, para tener una información más detallada se recomienda al lector revisar el artículo (Escorcia et al., 2013).
Modelado de los MCCs. Se utilizó la estructura cristalina de CalB (PDB 1TCA) (Uppenberg et al., 1995, 1994) a la que se le eliminaron dos moléculas de N-acetil-glucosamina unidas al residuo aminoacídico N74 debido a que se encuentran a una distancia de 18Å del grupo OH de la cadena lateral de la serina catalítica (S105). La verificación de la estructura y la adición de átomos de hidrógeno se realizó con PDB2PQR (Dolinsky et al., 2007; Dolinsky, Nielsen, McCammon, & Baker, 2004) y la propuesta de protonación se hizo con el programa PROPKA a pH 7.0 (Olsson, S0ndergaard, Rostkowski, & Jensen, 2011). Todos los residuos aminoacídicos potencialmente cargados fueron usados en su estado ionizado excepto el residuo D134. Posteriormente, el grupo hidroxilo de la cadena lateral de S105 se reemplazó por acetato para obtener la AcetilCalB y se etiquetó como SEA. Todas las aguas de cristalización fueron representadas por el modelo TIP3 (Jorgensen et al., 1983). La AcetilCalB se minimizó con los algoritmos Steepest Descent -SD- y adopted basis Newton-Raphson -ABNR- en una esfera de solvente explícito y se realizó una DM con el campo de fuerza CHARMM22 siguiendo un protocolo que involucra 12 resolvataciones de la proteína y calentamiento iniciando de 50 K a 300 K con aumento de 10 K cada 100 steps. Posterior al calentamiento se modeló la fase productiva de la DM durante 2ns. Una estructura representativa (con los enlaces de hidrógeno -a-, -b-, -d-, -e- y -f-menores a 4A) de la fase productiva se usó para realizar el acoplamiento molecular con el programa Autodock Vina (Trott & Olson, 2011). Para más detalles técnicos del procedimiento de solvatación, resolvatación, equili-bración y fase productiva de la DM se sugiere revisar la información adicional del artículo (Escorcia et al., 2013).
-Dinámica molecular QM/MM
Se calcularon seis trayectorias para los MMCs con los números de siembra (random seed) 55627, 1238, 384555, 835, 234 y 314159. Se utilizó un enfoque QM/MM y el método semiempírico Self Consistent Charge- Density Funtional Tight Bindig (SCC-DFTB) (Cui, Elstner, Kaxiras, Frauenheim, & Karplus, 2001; Elstner et al., 1998; Pu, Gao, & Truhlar, 2004) para el propranolol debido a la inexistencia de parámetros de campo de fuerza. Para la proteína (AcetilCalB), las aguas de cristalización y el solvente, tolueno explícito, se utilizó el campo de fuerza CHARMM22. Las moléculas de solvente se dispusieron en una esfera centrada en el C alfa de SEA105 con un radio de 40 Å. Solo una parte de la proteína podía moverse libremente, denominada región activa. La región activa incluyó todos los residuos aminoacídicos dentro de un radio de 30 Å. Los residuos aminoacídicos por fuera de este radio se mantuvieron fijos durante la DM. La esfera de solvente explícito se restringió con un potencial cuártico de frontera esférica iniciando con -0.25 kCal/mol a 39.5 Å, incrementándose a distancias mayores (parámetros: FORCE= 0.25, P1 = 2,25 y DROFF = 38.5). El algoritmo SHAKE se utilizó para restringir todas las distancias que involucraran átomos de hidrógeno (Ryckaert, Ciccotti, & Berendsen, 1977). Las interacciones electrostáticas entre las partículas dentro de un radio de 14 Å de la SEA105 se trataron de forma aditiva (Stote, States, & Karplus, 1991) y las interacciones electrostáticas a un radio mayor fueron aproximadas mediante el enfoque multipolo (Schlick, 2010). En la fase productiva se usó el algoritmo de integración de Verlet. El tiempo de simulación fue 2,5 ns, con un Time step de 1 fs. En total se modelaron 15 ns por cada MCC.
-Análisis de las trayectorias de dinámica molecular
En cada una de las trayectorias se visualizaron y analizaron 2,4 ns con el programa VMD (Humphrey, Dalke, & Schulten, 1996) (se descartaron los primeros 0,1 ns de la etapa de producción de la DM, asegurándonos que el sistema estuviera equilibrado). El análisis de los resultados fue dividido en dos secciones: evolución de los complejos de Michaelis y poblaciones de conformaciones de ataque cercano.
Evolución de los Complejos de Michaelis: En cada una de las DM se calculó el tiempo de vida de los MCCs y la desviación cuadrática media (RMSD) de las posiciones atómicas para todos los átomos pesados de la cadena principal de la proteína y los de los residuos aminoacídicos de las estructuras hélice alfas que rodean al sitio activo (figura 5). Estas fueron: i) Residuos L136 a T159 (hélice alfa 5, bucle, hélice alfa 6), ii) residuos A1 85 a V194 (lazo) y iii) L278 a V286 (segmento de la hélice alfa 10) (figura 1). También, se calculó la RMSD para los átomos pesados del (R,S)-propranolol.
Poblaciones de conformaciones de ataque cercano (NACs): Un NAC fue definido como un MCC en el que las distancias interatómicas -b- y -c- (figura 1) tienen un valor de < 2.7 A y de < 3.2 A simultáneamente (Escorcia et al., 2014). La población de NACs durante las trayectorias de DM se analizó y discriminó de acuerdo al modo de unión del (R,S)-propranolol en el sitio activo de AcetilCalB. Las poblaciones de NACs se reportaron como el tiempo de vida en ps.
RESULTADOS
Enlaces de hidrógeno del sitio activo de AcetilCalB relevantes para la actividad catalítica
La red de enlaces de hidrógeno -a-, -b-, -d-, -e- y -f- (figura 3) relevantes para la reacción de acilación del (R,5)-propranolol se conservó en todas las trayectorias de DM de los diferentes MCCs de (R,S)-propranolol en modo de unión I y II. El enlace de hidrógeno -a- que facilita la deslocalización de la carga en la histidina catalítica fue estable en todos los MCCs y trayectorias analizadas. Tuvo una distancia promedio de 1,9 Å y un ángulo D187O-H224H-H224N de 163°. Los tres enlaces de hidrógeno -d-, -e- y -f- (figura 3, tabla 1) que estabilizan al oxígeno carbonílico de SEA1 05 presentaron variaciones menores a 0,24 A, 0,18 A y 0,4 A respectivamente.
Tabla 1 Enlaces de hidrógeno SEA105O-HQ106 (-d-), SEA105O-HT4° (-e-) y SEA105O-HT4° (-f-) y ángulos SEA105O-T40H-T40N (-D-), SEA105O-T40H-T40N J-E-) y SEA105O-T40H-T40O de los complejos de Michaelis (MCC). Valores de las distancias n A y de los ángulos en grados (°). Entre paréntesis, la desviación estándar.
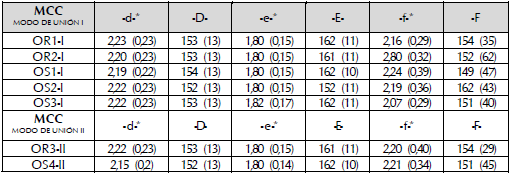
*Los enlaces de hidrógeno -d-, -e- y -f- (figura 2) estabilizan el O carbonílico de SEA105 du-rante el paso enantioselectivo de la reacción.
-Evolución de los MCCs y los NACs del (R,S)-propranolol con AcetilCalB
El enlace de hidrógeno -b- y la distancia -c- son un criterio para evaluar la aproximación del grupo OH del propranolol a la triada catalítica (figura 3). En el 70% de las 42 trayectorias analizadas, las distancias, -b- y -c-, fueron mayores a 4 A (tabla A1 en la información adicional) por lo que el tiempo de vida de los MCCs es apreciablemente menor que el tiempo de la simulación, indicando su baja estabilidad. Este resultado concuerda con la velocidad de la reacción determinada experimentalmente (Escorcia et al., 2013). El tiempo de vida de los MCCs y de los NACs (tabla 2) en cada una de las DM con las seis distribuciones de velocidades iniciales es notoriamente diferente (columnas i a la vi). De hecho, tanto el promedio del tiempo de vida como su desviación estándar presentan el mismo orden de magnitud (columnas: X y σ).
Tabla 2 Tiempo de vida (ps) de los complejos de Michaelis (MCCs) y de las conformaciones de ataque cercano (NACs) en cada una de las trayectorias de DM QM/MM durante los 2,4 ns finales de cada DM
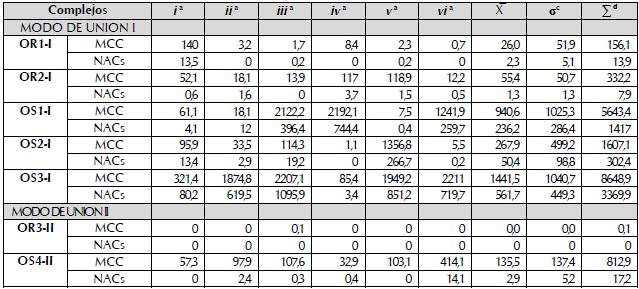
Números de siembra: i: 314159, i i: 835, i i i: 234, iv: 55627, v: 1238 y vi: 384555. b X: Población promedio de las seis trayectorias de DM QM/MM. ca: Desviación estándar. d S: Sumatoria del tiempo de vida de cada MCC en las trayectorias.
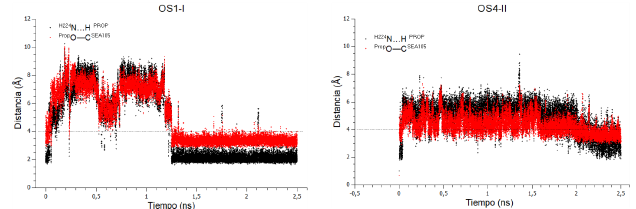
La sumatoria de los tiempos de vida de los MCCs y de los NACs por enantiómero y por modo de unión (tabla 3) indica que el tiempo de vida de los NACs con el S-propranolol en modo de unión I es ~ 352 veces mayor que en modo de unión II, y ~231 veces mayor que el de los NACs con el R-propranolol. Independiente del tipo de enantiómero, el tiempo de vida de los MCCs y de los NACs es menor en modo de unión II que en modo de unión I. A manera de ejemplo se muestra el comportamiento de las distancias que definen un NAC (distancias -b- y -c-) en las conformaciones OS3-I y OS4-II en fase productiva de la DM QM/MM con el número de siembra 38455, (figura A2, información adicional).
Tabla 3 Sumatoria de los tiempos de vida (ps) de los MCCs y de los NACs por enantiómero y por modo de unión.
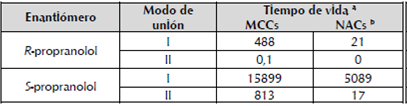
a Sumatoria del tiempo de vida en todas las trayectorias. b MCC con valores para el enlace de hidrógeno H224N--HPROP -b- y la distancia PRO" PO-CSEA105 -c- (figura 1) de < 2.7 A y < 3.2 simultáneamente.
DISCUSIÓN
Enlaces de hidrógeno del sitio activo de AcetilCalB relevantes para la actividad catalítica
La conservación de la red de enlaces de hidrógeno relevantes para la reacción de acilación del (R,5)- propranolol (tabla 1) en todas las trayectorias de DM de los diferentes MCCs de (R,S)-propranolol indica que la triada catalítica y el hueco oxianiónico de AcetilCalB se encuentran en una conformación estable para el proceso catalítico. Los enlaces de hidrógeno entre la SEA105 y el átomo de hidrógeno del enlace peptídico de Q1 06 -d- y T40 -f-, fueron más estables que el enlace de hidrógeno formado con el átomo de hidrógeno de la cadena lateral de T40 -e- (tabla 1). Esto sugiere una función importante en la estabilidad de SEA105. El modo de unión del (R,S)-propranolol al sitio activo de AcetilCalB no afecta la red de enlaces de hidrógeno relevantes para la actividad catalítica (figura 3).
-Evolución de los MCCs y los NACs del (R,S)-propranolol con AcetilCalB
Las diferencias poblacionales de los MCCs y NACs con los dos enantiómeros del propranolol, sugieren una enantiopreferencia por el S-propranolol en modo de unión I. Sin embargo, la formación de MCCs y NACs corresponden a etapas iniciales del proceso catalítico. Lo anterior indica que la enantioselectividad de la reacción se produce por diferencias en las conformaciones e interacciones repulsivas en el estado de transición (Ema, Kobayashi, Maeno, Sakai, & Utaka, 1998; Ema, Nakano, Yoshida, Kamata, & Sakai, 2012). Para corroborar estos resultados estamos realizando cálculos de las trayectorias del intermediario tetraédrico-2, así como de los perfiles de energía libre durante la reacción.
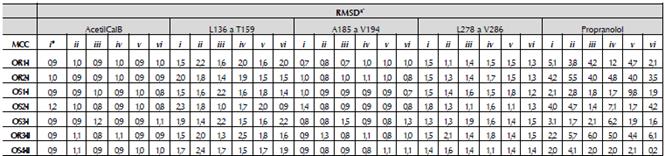
Las poblaciones de MCCs y NACs están relacionadas con variaciones en la longitud del enlace de hidrógeno -b- o con la distancia -c-. El aumento de la longitud estas distancias se debe a cambios conformaciones del (R,S)-propranolol y de la AcetilCalB. Los cambios conformacionales son mayores en el propranolol que en la Acetil-CalB (tabla 4 y tabla A3 en la información adicional). El valor promedio del RMSD de todos los átomos pesados del (R,S)-propranolol es aproximadamente cuatro veces más grande que el valor del RMSD de la cadena principal de la AcetilCalB o de las estructuras secundarias que rodean el sitio activo.
Tabla 4 Promedio de la desviación cuadrática media (RMSD) para los átomos pesados de la cadena principal de la AcetilCalB, y de las estructuras secundarias que rodean al sitio activo: i) Residuos aminoácidos L136 a T159, de la hélice alfa 5, bucle y hélice alfa 6. ii) Bucle compuesto por los residuos aminoacídicos A185 a V194. iii) Segmento de la hélice alfa 10, compuesta por los residuos aminoacídicos L278 a V286 y del R o S propranolol.
| RMSDa* | |||||
|---|---|---|---|---|---|
| Complejo | AcetilCalB | L136 a T159 | A185 aV194 | L278 a V286 | Propranolol |
| MODO DE UNIÓN I | |||||
| OR1-I | 0,95 | 1,82 | 0,87 | 1,38 | 5,32 |
| OR2-I | 0,93 | 1,68 | 0,95 | 1,45 | 4,33 |
| OS1-I | 0,93 | 1,68 | 0,95 | 1,45 | 4,33 |
| OS2-I | 0,93 | 1,68 | 0,95 | 1,45 | 4,33 |
| OS3-I | 0,92 | 1,58 | 0,88 | 1,50 | 3,35 |
| MODO DE UNIÓN II | |||||
| OR3-II | 0,92 | 1,58 | 0,88 | 1,50 | 3,35 |
| OS4-II | 0,92 | 1,58 | 0,88 | 1,50 | 3,35 |
a Calculado usando como referencia la estructura cristalina de CalB (1TCA) (17) y el plugin RMSDTT v3.0 para el programa VMD (33).
http://physiology.med.cornell.edu/faculty/hweinstein/vmdplugins/rmsdtt/index.html
* El valor promedio se calculó teniendo en cuenta todas las trayectorias.
A diferencia del trabajo reportado con anterioridad, nuestro análisis se centró en el tiempo en el que la conformación de ataque cercano se mantuvo a lo largo de l a trayectoria y no en el porcentaje de NACs presentes en la población de MCC reactivos. Los resultados de cada una de las réplicas fueron notoriamente diferentes como lo indican los valores del promedio y de la desviación estándar (tabla 2). Esto nos permitió evaluar el efecto de la modificación de los números de siembra sobre las poblaciones de MCC reactivos y NACs. Las poblaciones de MCC reactivos y de NACs para el complejo OS1-I se muestran a manera de ejemplo (figura 6).
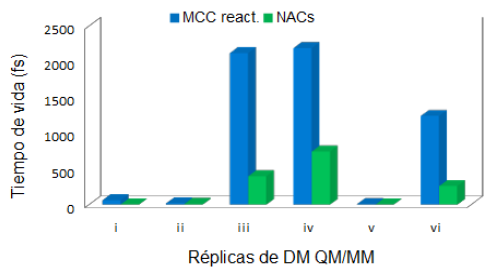
CONCLUSIONES
En este trabajo presentamos un estudio detallado del efecto del cambio en la distribución de velocidades iniciales asignadas a los átomos durante la dinámica molecular sobre la formación y evolución de los complejos de Michaelis (MCCs) y de las conformaciones de ataque cercano (NACs) en la reacción de acilación del (R,S)-propranolol catalizada por CalB. Nuestros resultados indican que:
Las poblaciones de los MCCs y de los NACs son dependientes de la distribución de las velocidades iniciales de la dinámica molecular.
Los MCCs del S-propranolol y CalB son más estables que los MCCs con el R-propranolol. Es decir que el S-propranolol está favorecido al inicio del paso enantioselectivo de la reacción de desacilación de AcetilCalB.
Los MCCs y los NACs presentan un tiempo de vida mayor cuando el propranolol se orienta en el sitio activo de CalB en modo de unión I que en modo de unión II.
La enantioselectividad moderada de AcetilCalB, encontrada experimentalmente, puede ser parcialmente atribuida a la alta población de NACs observados para el S-propranolol. Se sugiere estudiar las trayectorias del intermediario tetraédrico-2, así como los perfiles de energía libre durante la reacción de desacilación de Acetil-CalB.