










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista U.D.C.A Actualidad & Divulgación Científica
Print version ISSN 0123-4226
rev.udcaactual.divulg.cient. vol.15 no.2 Bogotá July/Dec. 2012
CIENCIAS EXACTAS Y NATURALES - Artículo Técnico
ION MOBILITY SPECTROMETRY: HISTORY, CHARACTERISTICS AND APPLICATIONS
ESPECTROMETRÍA DE MOVILIDAD IÓNICA: HISTORIA, CARACTERÍSTICAS Y APLICACIONES
Roberto Fernández-Maestre1
1 Licenciado en Biología y Química, Doctor en Química Analítica, Docente del Programa de Química, Campus de Zaragocilla, Universidad de Cartagena, Cartagena, Colombia. rfernandezm@unicartagena.edu.co
Rev. U.D.C.A Act. & Div. Cient. 15(2): 467 - 479, 2012
SUMMARY
Ion mobility spectrometry (IMS) is an analytical technique that separates ions in the gas phase. Ions are separated at atmospheric pressure under the influence of an electric field, according to their size and shape. IMS is the best choice for detection of narcotics, chemical and biological warfare agents and explosives in airports and customs. IMS can detect almost anything that can be ionized and has been applied to the analysis from the lightest elements such as helium to the most complex mixtures such as proteomes, metabolomes and complete organisms such as bacteria, chiral separations, and structure determination. Although since 2000 there have been approximately fifty reviews of IMS, this review is probably the only general valuation of this technique since then.
Key words: Ion mobility spectrometry, gas-phase ions, reduced mobility, ion sources.
RESUMEN
La espectrometría de movilidad iónica (IMS) es una técnica analítica que separa iones en fase gaseosa. Los iones son separados a presión atmosférica bajo la influencia de un campo eléctrico de acuerdo a su tamaño y forma. IMS es la mejor opción para detectar narcóticos, agentes químicos y biológicos de guerra, y explosivos en aeropuertos y aduanas. IMS puede detectar casi cualquier cosa que pueda ser ionizada y se ha aplicado al análisis de elementos ligeros como el helio, mezclas más complejas como proteomas, metabolomas y organismos completos, tales como bacterias, separaciones quirales, y la determinación de estructuras. Aunque desde el año 2000 se han presentado aproximadamente cincuenta revisiones de la IMS, la presente es probablemente la única en evaluar este tema de manera general desde esa fecha y es probablemente la primera en publicarse en una revista latinoamericana.
Palabras clave: Espectrometría de movilidad iónica, iones gaseosos, movilidad reducida, fuentes de ionización.
INTRODUCTION
Ion mobility spectrometry (IMS) is an atmospheric pressure technique for trace analysis of gas-phase analytes. IMS separates ions in an electric field in the presence of an inert gas on the basis of their mobilities, a measure of the size- to-charge ratio of an ion. IMS can be used for selective detection of ions after a chromatographic separation, for pre- separation of ions before mass spectrometry or, as a stand- alone instrument. Ions of organic or inorganic compounds, elements, particles and organisms can be detected. IMS is especially sensitive to organic compounds such as illicit drugs, chemical and biological warfare agents and explosives. Analysis can be carried out in a matter of seconds; this is the reason why IMS is the technique of choice to detect these materials at customs and in airports and has a wide use in military applications.
History: Ernest Rutherford measured the mobility of ions formed by x-ray ionization (1897) and characterized the ions using ion mobilities (1899). During the first three decades of the 20th century, there was a strong interest in mobility studies and a large body of theory on ion kinetics and experimental data was compiled. In that period, the effect of collisions, attractive forces, temperature, pressure, accelerating voltage, and contamination on mobilities were recognized (Langevin, 1903).
In the 30's and 40's, the interest for ion mobility declined due the introduction of mass spectrometry, which was free of the complicated reactions present at the pressures used for mobility studies. The period 1948-1970 has been mentioned as foundational studies (Eiceman & Karpas, 2005); a number of theoretical studies in ion mobility by Mason & Schamp (1958) and McDaniel (1964) were conducted in this period, creating the base of modern IMS. In this epoch, there was a renewed interest in mobility studies made known by: a) primitive ion detectors, used by military forces during and after world word II for the detection of fuel from submarines, and other applications (Eiceman & Karpas, 2005); b) an ionization anemometer, invented by Lovelock in 1948, that was sensitive to organic vapors (Lovelock & Wasilewska, 1949) which opened the possibility of using mobility instruments for chemical analysis; and c) the construction of suitable drift tubes, such as that of Albritton and McDaniel, similar to modern drift tubes (Albritton et al. 1968).
IMS was introduced as an analytical tool by Cohen & Karasek (1970). In an ion mobility spectrometer, organic molecules are ionized and driven by an electric field against a counterflow of neutral drift gas. In their way to the detector, the ions collide multiple times with the drift gas, which reduce their speed. After each collision, ions are accelerated again by the imposed field. The alternation of accelerations and collisions results in a constant average ion velocity that depends on the ion charge, mass, and collision cross section. This dependence allows the identification of the ions by their arrival time at a downfield detector (St. Louis et al. 1989).
The second edition of a book on IMS is available (Eiceman &Karpas, 2005) and several IMS reviews have been published. Márquez-Sillero et al. (2011) assessed environmental applications of IMS, the analytical tools developed to solve the limitations regarding selectivity and sensitivity and its coupling to other detection systems; IMS coupled to gas chromatography for the sensitive and selective detection of compounds after chromatographic separation was reviewed by Kanu &Hill (2008); gas chromatography-IMS has proved versatile for the sensitive and selective detection of compounds, especially complex mixtures in difficult matrices; Kanu et al. (2008) compared and contrasted several types of ion mobility-mass spectrometers and described their advantages for application to a wide range of analytes; Johnson et al. (2007) evaluated IMS potentials in space exploration including IMS in manned space flight, the International Space Station Volatile Organic Analyzer, IMS in robotic space exploration, potential extraterrestrial missions and current/future directions and development. Finally, Weis (2005) reviewed IMS in combination with quantum chemical calculations to determine the structure of cluster ions of metals and semi-metals; they found that clusters of less than 100 atoms show a rich variation in shape as function of the number of atoms. Other review focused on ion sources (Guharay et al. 2008) and most others on the study of large macromolecule interactions and structure elucidation.
IMS main advantages and disadvantages are presented in table 1.
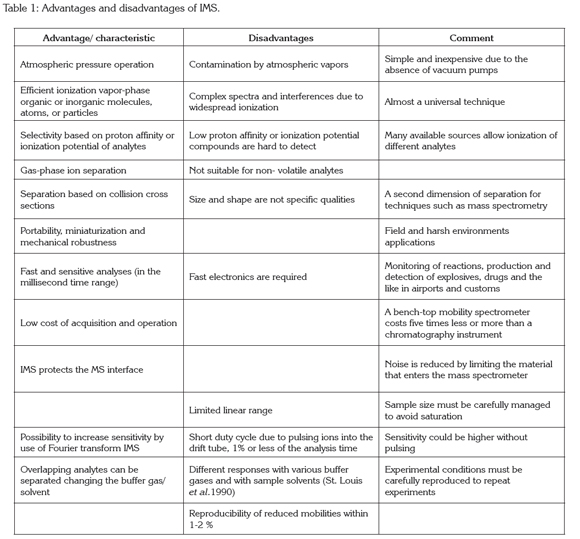
Instrumentation: The ion mobility spectrometer consists of three basic units kept at atmospheric pressure: an ionization source and an ion drift tube (Figure 1) maintained at either a positive or at a negative uniform electric field gradient, and a detector. Ions produced in the ionization source are accelerated down the electric field where they are separated according to their mobilities in a countercurrent flow of inert gas.
Ion sources: Ionization methods to convert molecules into ions to be separated in the drift tube in IMS include 63Ni b ionization (Cohen & Karasek, 1970), photoionization (Baim et al. 1983), laser ionization (Lubman & Kronick, 1982a), corona spray ionization (Tabrizchi & Rouholahnejad, 2004), electrospray ionization (McMinn et al. 1990), and other sources (Gunzer et al. 2010). The most used are 63Ni b and electrospray ion sources.
Vapors of analyte in the ionization region are ionized directly (MALDI, UV, and laser ionization) or by reaction of the analyte with reactant ions produced by the ionization source through a series of charge transfer reactions (63Ni b, corona discharge, chemical, and electrospray ionization). The reactant ion is (H2O)nH+ when dry nitrogen (5-10 ppm of H2O) is used as the drift gas, where n is 1-4 depending on the moisture and temperature; when air is used as the drift gas, (H2O)nH+ and (H2O)nO2 - or (H2O)n(CO2)mO2- are the reactant ions for positive and negative ion detection, respectively (Hill &Simpson, 1997).
Radioactive sources: The 63Ni foil, the ion source of the first mobility spectrometers, is a secondary ionization source analogous to that found in an electron capture detector. Ionization in this source is produced by the emission of electrons from the radioactive source with average energies of 19 keV. These electrons collide with neutral molecules of analyte or drift gas and ionize them by a series of charge transfer reactions. 63Ni response is nonlinear, and like other charge transfer ionization sources (corona discharge and chemical ionization), reactant ions can undergo interfering reactions with contaminating compounds. These contaminating compounds include chromatographic column bleed compounds and components in the sample mixture. As reactant ions are depleted by these competing reactions, response to the compound of interest becomes erratic or is eliminated (Baim et al. 1983). Other radioactive isotope less frequently used is 241Am, that emits more energetic electrons that can exceed 5 MeV (Guharay et al. 2008). An advantage of radioactive sources is that they do not require a power supply and, consequently, are suitable for portable instruments. Disadvantages are radioactive contamination due to wrong manipulation, the need to supply the samples in vapor phase, and bureaucratic complications due to governmental regulations.
Electrospray Ionization: The development of electrospray ionization (ESI) was successfully introduced to IMS by Hill and greatly expanded the range of compounds that could be analyzed by IMS (Hallen et al. 1989). In the ESI process, a high electric potential is applied to the needle of the sample injection syringe, which creates electric charges. Electrospray occurs when the sample liquid is drawn by a coulombic force from the needle toward the target electrode (target screen, Figure 2) that is held at a lower voltage (~3.5 kV). As it travels toward the target electrode, solvent evaporates leaving increasingly charged droplets that 'explode' due to coulombic repulsion. This process produces droplets of increasingly smaller radius, ideally culminating in molecular ions (De Hoffmann & Stroobant, 2001). Electrospray sources are ideal for liquid samples and non-volatile high molecular weight analytes. Electrospray is a soft ionization source that yields simple spectra with no fragmentation where the molecular weight can be easily determined when coupling IMS to mass spectrometry. The use of electrospray IMS as a separation and detection device has been demonstrated for explosives (Asbury et al. 2000), chemical warfare degradation products (Rearden & Harrington, 2005), and biological mixtures (Valentine et al. 1998).

Secondary electrospray ionization: (SESI) was first introduced to IMS by Hill in 2000 (Wu et al. 2000). In SESI, a usual ESI device produces solvent ions that, acting as reactant ions, ionize liquid or gaseous analytes. SESI-IMS-MS has been applied to the detection of illicit drugs, where it was found to be more sensitive than ESI-IMS-MS (Wu et al. 2000). SESI allows easy and fast sampling by applying jets of ions with a probe and picking up the secondary ions with a second probe to specific sites on a surface; therefore, SESI can sample difficult-to-access surfaces, organelles on a cell and map and image surfaces. SESI also allows semi non-destructive analysis to evaluate valuable objects since the jet of ions exerts negligible damage to sampled objects.
Corona-spray and corona-discharge ionization: In corona ionization, a high electric field is applied to the electrospray needle tip and the bath gas surrounding the needle ionizes. These ions react with neutral molecules, which may evaporate from the liquid at the needle tip. Applications of corona ionization include O2 generation (Sabo & Matejcik, 2011) and detection of volatile organic compound (Boggio et al. 2011). Corona -spray and corona discharge are alternatives to conventional radioactive ionization; the high power consumption of DC corona discharge becomes one of the limits to usefulness in portable IMS systems for which a short pulsed corona discharge source has been developed (Yuan et al. 2005).
Matrix-assisted laser desorption ionization (MALDI): MALDI was first coupled to IMS by 1990 (Wyttenbach et al. 1996). In MALDI, macromolecules such as proteins or DNA strands are dissolved in a solution of a small organic molecule (matrix). The solution is dried on a target and a laser pulse is applied. The matrix absorbs the laser pulse and sublimes carrying some analyte. Singly charged protonated molecules are produced during the sublimation or in the gas phase, making the analysis simpler and potentially more sensitive (De Hoffmann & Stroobant, 2001). MALDI is ideal for the determination of molecular weights and analysis of macromolecules since it does not fragment analytes.
Photoionization sources: These sources use photoionization lamps and lasers. Photoionization is achieved through the use of a short wavelength UV lamp and is an inexpensive, practical alternative to laser sources for use in an ion mobility GC detector for aromatic and other unsaturated organic compounds; their major advantage is that, by adjusting the wavelength, the analyst can selectively ionize predetermined compounds; other advantages includes the lack of reactant ions enabling the use of the entire Ion mobility spectrum from 0 to 20m for observation of analyte Ions (Baim et al. 1983). Ultraviolet light from a NdYAG pulsed and ArF excimer lasers was used by Lubman & Kronick (1982b) at atmospheric pressure as ionization sources in an ion mobility spectrometer; they found advantages such as the production of only one peak, the molecular ion or MH+, reducing the problem of multiple peaks occurring in IMS, great sensitivity, i.e., at least down to 1 ppb for benzene, and additional means of discrimination by the use of a particular wavelength. Disadvantages of UV lamps are the moderate energies supplied that limit the ionization and the types of compounds analyzed.
Drift tube: In the drift tube, ions are separated by an electric field before entering the detector. The following description corresponds to a traditional drift tube: the drift tube is usually made of a series of stainless-steel guard rings between insulating quartz, glass, or ceramic rings (99.5% Al2O3), stacked on top of one another to form a completely enclosed tube (Figure 1). Each guard ring is connected to the next one in series through 1-MΩ or 0.5 MΩ resistors (Figure 2). A high electrical potential (~12 kV) is placed on the first guard ring, the target screen, to produce a 200-400 V/cm field throughout the drift tube (Fernández-Maestre et al. 2010a); alteration of the length of the ion separation region by addition or removal of stainless steel rings is possible. The rings are held inside a ceramic tube (Figure 2b) that has an aperture all along its length to introduce the electric contacts of the rings; this ceramic tube is housed inside an aluminum oven for heating (Figure 2a). Previous IMS designs used round insulator beads, which produced large apertures between the guard rings; this open design allowed undesired neutral species or radicals in the tube; the introduction of the close design.
Ion gates: Once the gas-phase ions are formed in the ionization source, they are directed by the electric field down the drift tube toward the detector. On their way, they encounter sets of parallel wires that prevent the ions from continuing their migration through the spectrometer. These sets of parallel wires are called ion gates. The entrance ion gate is placed at the beginning of the drift region and is electronically opened for a few tenths of a millisecond to permit a pulse of analyte ions to enter this region; typical pulses are 0.2m long. The gate is open (all ions pass) when each gate wire is at the potential of the drift field at that place in the drift tube and is closed (ions are stopped) when a potential higher than the drift voltage is placed between each pair of adjacent wires. After passing the gate, the ions drift with the electric field, some faster and some slower according to their individual ion mobilities, and arrive at the collector electrode at different times. Before arriving to the detector, ions can find a second gate placed just in front of it. The purpose of this aperture grid is to shield some detectors from the inductive effects of the incoming ion cloud. With no aperture grid, a collector electrode responds to the ion cloud before the cloud arrives at the electrode, producing a broadened ion peak (Hill & Simpson, 1997). The aperture grid can be opened at progressively larger intervals after the entrance gate to create an ion mobility spectrum or can be opened at fixed intervals after that gate to monitor only ions of a given mobility (Baim & Hill, 1982). The use of ion gates decrease the sensitivity since they are open only a fraction of the analysis time; to increase sensitivity, Fourier Transform IMS (FTIMS) and pulsed sources have been used; in FTIMS, a second ion gate is placed close to the collector, and synchronized with the entrance gate at a rate that is continuously varied from low to high frequency. As ions migrating through the drift region of the spectrometer go in and out of phase with the oscillating gates, the ion current at the collector increases and decreases producing an interferogram; signal to noise ratio increase in FTIMS because he gates are open 50% of the time (Hill & Simpson, 1997; Eatherton et al. 1988).
Drift gases: A countercurrent of dry neutral gas is used in IMS as a clean and inert matrix through which ions drift. The drift gas also serves to keep the spectrometer drift tube clean by keeping neutral compounds, introduced with the sample or coming from the atmosphere, from passing into the drift region (Hill & Simpson, 1997). The drift gas, usually nitrogen or air, enters the bottom of the spectrometer with flow rates on the order of 0.5-1.5L/min, passes through the drift tube and exits through the ionization region (Figure 1, buffer gas entrance). Helium, carbon dioxide, and argon also have been used as drift gases; when drift gases are changed, sensitivity and resolving power change; helium was by far the most sensitive, giving nearly nine times more peak area than that seen in nitrogen; fast ions have lower resolving powers due to increased contributions from the ion pulse width (~0,2m) to the overall peak width, whereas for slower drifting ions, diffusion becomes the main band-spreading mechanism; when the effect of the starting pulse width was removed, the drift gases all performed nearly identically, which indicates that drift gases produce similar resolving powers (Asbury & Hill, 2000).
Carbon dioxide has been used as a drift gas when coupling IMS to supercritical fluid chromatography (SFC). Use of carbon dioxide as a drift gas was difficult in earlier IMS experiments because it formed such large clusters with ions that the mobility of the ion cluster was independent of the core ion species (Ellis et al. 1976); however, when analyzing large molecules or using temperatures higher than 220°C this situation changes; it was demonstrated that the patterns of the ion mobility spectra were similar to those for nitrogen while ion drift times were considerably longer in CO2; unfortunately, these longer drift times lead to broadening by diffusion and reduced sensitivity (Rokushika et al. 1987); also, when using unidirectional flow FTIMS as a detector for SFC and nitrogen as a drift gas, there were no differences in the mobilities of the reactant ions caused by CO2 contamination, indicating that the identities of the reactant ions were unaffected by CO2 flow (Eatherton et al. 1988) maybe because the high temperature of the IMS tube did not allow clustering with CO2; for flows above 40mL/min, the signal for reactant ions decreased and eventually disappeared, which makes necessary to split the the chromatographic flow for packed columns (Morrissey & Widmer, 1991).
Doping agents: Doping agents (reagent gases) added to the drift gas control ionization and increase selectivity in IMS. When using methylene chloride as a doping agent for the detection of explosives, the negative ion Cl2 selectively attaches these electronegative molecules and sensitivity increase (Lawrence & Neudorfl, 1988). In the positive ion mode, low proton affinities compounds such as normal hydrocarbons are unresponsive. To observe hydrocarbon signal, water must be purged from sample and drift gas, and a dopant agent must be added to the drift gas or a metastable helium ionization source must be used; in this source, an inert gas in the presence of a strong electric field can be excited to a metastable state through collisions with electrons from a β-source; this excited molecules can then ionize molecules with high ionization potentials (Kojiro et al. 1991). The addition of doping agents to ion mobility spectrometers to selectively ionize compounds was first applied by Kim et al. (1978) who added ammonia to the N2 buffer gas to ionize amines. When using a very high affinity compound like NH3 as a doping agent, the selectivity increases since fewer compounds compete with ammonia for proton and only very strong gas-phase bases are detected, for example amines and drugs (Kim et al. 1978). Other doping agents used to ease analysis are carbon tetrachloride for explosives (Spangler et al. 1985) and dichloromethane, dibromomethane, methyl iodide, acetic acid, dimethyl sulfide and acetonitrile for explosives (Proctor & Todd, 1984).
During the analysis of high proton affinity vapor analytes with IMS in air, a high number of interferences arises due to the small proton affinity of water; one way to circumvent this is to add the drift gas with small quantities of ketones, which allows the formation of dimers with a higher stability than that of water clusters. The spectra will simplify because only compounds whose proton affinities are above that of the acetone dimers are detected (Hill & Simpson, 1997). Other examples of the addition of doping agents to the buffer gas are acetone and dimethylsulfoxide added to mixtures of volatile organic and organophosphorus compounds (Eiceman et al. 1995); acetone, water, and dimethylsulfoxide added to volatile organic compounds (Meng et al. 1995); acetone and 5-nonanone added to hydrazine and monomethylhydrazine to skip the interference of ammonia (Eiceman et al. 1993); 4-heptanone added to alkanolamines in the presence of interferences of ammonia, Freon 22, and diesel fuel (Gan & Corino, 2000), and ketones added to hydrazines to avoid ammonia interference (Bollan et al. 2007). The use of doping agents in IMS was reviewed by Puton et al. (2008). All these researchers introduced the doping agents to the buffer gas in the reaction region of the mobility spectrometer to selectively change ion mobilities but only to avoid interferences.
The application of these selective changes in ion mobilities due to addition of a doping agent to the buffer gas had been applied to separate interferences but not to separate analytes with similar K0 values. Doping agents that are introduced by the end of the drift region, and not with the analyte in the reaction region, are called buffer gas modifiers. Separation is achieved because selective clustering of the modifiers with the analytes occur which change analyte mobilities depending on the size and steric hindrance on the charge of the analyte ions. Sugar, drug and amino acids enantiomers were separated using (S)-2-butanol (Dwivedi et al. 2006); however, these authors only used the differences between enantiomers and did not take advantage of the differences in compounds mobilities. Fernández-Maestre et al. (2010b) did so by separating overlapping α-amino acids using 2-butanol and demonstrated the formation of analyte- modifier clusters; the authors also observed a decrease on cluster formation with temperature increase. The formation, or the lack of formation, of clusters analyte-modifiers was also demonstrated for 2,4-lutidine, 2,6-di-tert-butyl pyridine, tetraalkylammonium ions, and a-amino acids using water, methyl 2-chloropropionate, and trifluoromethyl benzyl alcohol as modifiers (Fernández-Maestre et al. 2010a). The formation of gas-phase intramolecular bridges in diamines such as arginine, histidine, and lysine and the drug atenolol was demonstrated by Fernández-Maestre et al. (2012) introducing modifiers in the buffer gas on a mobility spectrometer. The diamines mobilities were unaffected when modifiers were introduced into the buffer gas due to the formation of intramolecular bridges that hindered the attachment of modifier molecules to the positive charge of ions and delocalized the charge, which deterred clustering; ethyl lactate, nitrobenzene, 2-butanol, and tetrahydro-furan- 2-carbonitrile were used as modifiers. Separations with the addition of buffer gas modifiers opens up prospects for extending the application of IMS to the determination of complex mixtures.
Detection methods: In IMS, the most common and simple detection device to measure the ion intensity is a collector plate that works as a Faraday cup. In many instruments, a biased aperture gate placed close to this cup serves either as a detector or to increment the efficiency of the Faraday cup. This gate prevents the buildup of an ion charge on the collector plate, imparts energy to the ions to increase collection efficiency and filters out artifact signals coming from the opening and closing of the entrance gate (Eiceman & Karpas, 2005). Faraday cups can be replaced by detectors such as mass spectrometers that introduce additional identification capabilities to IMS.
Ion Mobility-mass spectrometry (IMS-MS). A common detection technique for IMS is mass spectrometry (MS). Coupling MS to IMS allows the determination of molecular weights, fragmentations, clustering and other type of reactions in the drift tube. All kinds of MS instruments have been interfaced to IMS systems including quadrupole (Clowers & Hill, 2005), time of flight (Ugarov et al. 2004), Fourier-transform ion cyclotron resonance, FTICR (Bluhm et al. 2000), sectors (McDaniel et al. 1962) and ion trap (Clowers & Hill, 2005). The coupling of these detectors to IMS adds an identification dimension to the detection of ions. When coupling IMS to MS, there are several possible modes of operation. In the IMS only mode, MS serves as a detection technique for IMS and no scan is performed: all ions reach the detector without scanning and the resulting spectra are similar to those in IMS alone operation; the drift time change is negligible because, although the distance the ions travel inside the MS instrument is similar to that in IMS, in MS the ions travel much faster due to the absence of drift gas in the vacuum conditions. In the MS mode, both gates are open (in the case of quadrupoles) and all ions pass continuously and directly to the mass spectrometer and are mass analyzed; the mass spectrum is obtained in this mode. In the case of IMS-tof-MS, the ions are sent by pulses to the MS. In the IMS- MS mode, all peaks of the IMS spectrum are mass analyzed continuously; this is possible because one mass spectrum can be acquired in less than one millisecond, so several MS spectra can be taken for every mobility peak; therefore, separation of all the ions with different masses and the same mobilities can be separated. In Selected Ion Monitoring mode, the MS instrument is set to detect one determined mass-to-charge value; the result is an ion mobility spectrum of all ions with the specified mass.
IMS analysis: Sample introduction: Samples can be introduced in the IMS tube directly if they are in a vapor form. Liquid samples or solutions can be introduced by means of ESI, SESI, and corona spray ionization sources, or through a chromatographic instrument, and solid samples may use MALDI and thermal or laser desorption. Direct injection the environment and chloride and nitrate ions limit the capability of IMS to analyze compounds with low proton or electron affinities and introduce other unwanted reactions. Membrane inlets allow sample introduction that keep reactive molecules such as water and ammonia out of the reaction region; membranes are common when non-purified air is used as the buffer gas such as in field measurements. Spangler & Carrico (1983) tested two membranes for sample introduction: MEM-100 dimethylsilicone and Celgard 2400 (microporous polypropylene film); non-porous membranes as dimethylsilicone were capable of reducing significantly all atmospheric contaminants except CO2; polypropylene membranes were less effective to diminish intake of reactive molecules from sampled air.
Introducing the sample using a chromatographic technique gives a second dimension for easy resolving complex mixtures. Gas chromatography (GC) was first coupled to IMS by Karasek (1970) and later was liquid chromatography, SFC, and capillary electrophoresis (Hallen et al. 1989). SFC-IMS has been applied to the determination of nicotine in tobacco (Wu et al. 1998), GC-MS for the analysis of bacteria by pyrolysis (Dworzanski et al. 2005), and liquid chromatography for analysis of carbohydrates (Lee et al. 1998).
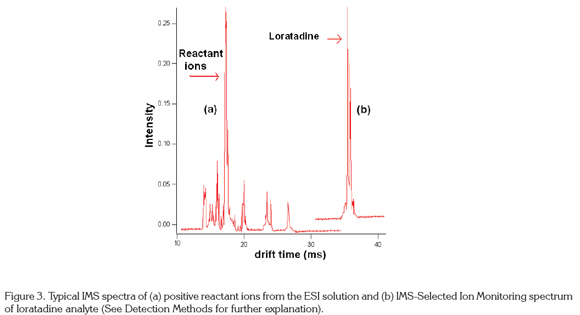
Spectra: Spectra are graphs of intensity of the ion peaks (ion current) vs. drift time on the x axis. The zero drift time is taken as the time the entrance gate is open (in occasions when the gate is closed) to let the ions enter the drift tube. Most of the ions are neutralized in the aperture gate, which is open only about 1% of the duty cycle. Signals are very noisy and it is necessary to average as many as 250 spectra to obtain a clean spectrum. However, the total analysis time is less than one minute, faster than most other separation techniques (Eiceman & Karpas, 2005). Figure 3a shows a typical ion mobility spectrum of the positive reactant ions in nitrogen, in this case, the water reactant ion, (H2O) H+. IMS spectra can show drift times of 10m for small molecules to more than 100 ms for macromolecules like proteins and nucleic acids, being the most common about 20-25m (Hill & Simpson, 1997). Figure 3b shows a loratadine IMS spectrum with a drift time of 35m. In 2-D mode, the drift time is displayed in the y axis of the spectrum vs. m/z in the x axis. This mode is useful to study complicated spectra such as proteomes (McLean et al. 2005) and metabolomes (Dwivedi et al. 2010).
Reduced mobility: Ion mobility spectrometry separates compounds on the basis of their different gas-phase velocities, commonly in nitrogen or air, giving mobility constants (K), defined as:

Where v is the velocity of the ion in cm/s and E is the electric field in the drift region of the spectrometer in V/cm. If weakfield conditions exist (i.e. less than 500 V/cm), v should be a linear function of E. Mobility constants are more easily calculated by measuring the time an ion travels down the drift tube. Mobilities can then be found by replacing E = V/L and v = td/L in eq. 1

Where L is the distance the ion travels to reach the detector in centimeters, V is the total voltage drop in the drift tube that forces the ions through the drift tube in volts, and td is the time in seconds the ion takes to travel down the drift tube till the detector. To compare ion mobilities in different experimental conditions mobilities must be normalized to standard conditions obtaining the reduced mobility (in cm2 V-1 s-1):
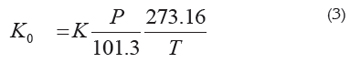
Where P is the pressure in kPa and T the temperature in K. Factors affecting Ion mobilities at atmospheric pressure comprise mass, size, and charge (Revercomb &Mason, 1975). Reduced mobilities exhibit a mass-related temperature dependence for homologous series of compounds e.g., alcohols, carbohydrates, amines; for amines, this temperature dependence is positive (i.e., mobility increases with increasing temperature) for low molecular mass ions, almost constant for intermediate masses (90-180 Da) ions, and negative for heavy ions (Berant & Karpas, 1989). Reduced mobility values are reproducible to within 1-2% in different laboratories (Eiceman & Karpas, 2005); a compilation of these values has been published (Shumate et al. 1986).
Calibration: To calibrate the IMS instrument, the fact that under certain conditions the product K0t is constant can be used. This means that the reduced mobility of different analyte ions may be calculated from that of a calibrant, K0c:
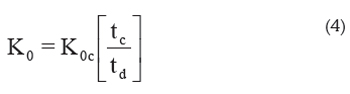
where tc is the drift time of the calibrant at the specific conditions of the experiment and td is the drift time of the analyte at the same conditions (Eiceman et al. 2003). 2,4-lutidine is the compound commonly chosen as calibrant (Berant & Karpas, 1989). This method skips the reading of barometers and eliminates the errors due to wrong measurements of the parameters in eq. 3 but can introduce errors due to contamination of the drift tube which can produce clustering that change ion mobilities.
Tetraalkylammonium ions, 2-4, lutidine, and di-tert- butylpyridine are good standards for IMS because they produce a single peak and a very sensitive signal. Fernandez- Maestre et al. (2010a) addressed issues such as errors produced by contamination and demonstrated that 2,4-lutidine or the single use of one calibration standard could lead to errors when contamination was present; they proposed the use of a standard with a mobility affected by temperature or contamination, such as 2-4, lutidine, to detect contamination, followed by an standard unaffected by temperature or contamination (due to its low clustering tendency, such as a tetraalkylammonium salt), to be used in eq. 4. Linear calibration in IMS ranges from 10 to 1,000mM. The reactant ions are consumed proportionally to the analyte concentration and their signal decrease and that of the analyte ions increase with the concentration of the analyte. At high concentrations, analyte dimers appear and the analyte eventually consumes all available reactant ions and its signal reaches a maximum; this situation should be avoided because the quantitation characteristics of the instrument are lost; therefore, reactants ions should be always visible in the spectra (Eiceman & Karpas, 2005).
Collision cross sections: Kinetic theory yields a form for the mobility at standard temperature and pressure conditions (Mason & Schamp, 1958):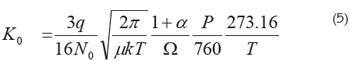
In this equation, (the Mason-Shamp equation), P is the pressure in Torr., T the temperature in °C, q the charge of the ion, N0 the gas number density at standard temperature and pressure conditions (N0 = P/kT), μ the reduced mass of an ion-drift gas pair, k the Boltzmann constant, ω the ionneutral collision cross section and α a small correction term with a magnitude of less than 0.02, when the ion mass is larger than the mass of the drift gas molecule. The reduced mass is defined as mM/(m + M) where m and M are the molecular mass of the analyte and the drift gas, respectively; this equation is useful for calculating collision cross sections of molecules, especially important for macromolecules like proteins that can adopt different conformations, which are closely related to their biological activity.
Applications of IMS: Only in this decade, commercial bench top IMS analyzers were available after decades of absence from the market. The GA-2100 Electrospray IMS is commercialized by Excellims Corporation (2011) as being "faster than HPLC... perfect for rapidly and sensitively analyzing liquid samples... Pharmaceutical Cleaning Validation, process monitoring, and many other types of analysis". IMS has had dissimilar applications such as the detection of contaminants in food (Bota & Harrington, 2006), analysis of protein structures (Myung et al. 2006), determination of illegal drugs in human hair (Sheibania et al. 2011), detection of moulds (Tiebe et al. 2010), identification of wood species (Lawrence et al. 1991) and detection of emissions from surfaces (Vautz et al. 2006), and has been proposed as an analytical separation tool for searching the chemical signatures of life during exploration of solar system bodies (Johnson et al. 2007).
In biology, IMS has been applied to the determination of bacteria by enzyme substrate reactions. A method add (o-nitrophenyl) galactopyranoside to cell cultures where bacterial enzymes cleave it to o-nitrophenol; this relatively high vapor pressure product can be detected by IMS sampling the headspace of the sample (Snyder et al. 1991). Applications in medicine include the detection of drugs in breath of patients (Carstens et al. 2010), determination of methanol and ethanol in human saliva (Bocos-Bintintan et al. 2010), and volatile metabolites to diagnose chronic obstructive pulmonary disease (Bessa et al. 2011), bronchial carcinoma (Finthammer et al. 2010), and other diseases (Bunkowski et al. 2010). IMS has also been applied to the detection of acetaminophen, aspartame, bisacodyl, caffeine, dextromethorphan, diphenhydramine, famotidine, glucosamine, guaifenesin, loratadine, niacin, phenylephrine, pyridoxine, thiamin, and tetrahydrozoline in over-the-counter-drugs and aspartame and caffeine in beverages (Fernandez- Maestre & Hill, 2009). These applications could be of interest in third-world countries due to the low cost of this technique, especially for countries like Colombia where cheap and sensitive methods of medical diagnostic are required.
Not only are organic compounds detected by IMS but airborne molecular contamination (Shupp et al. 2007) and many inorganic species can be monitored continuously; these inorganic species include as Cl- , l- , Br- , HF, HCl, HI, HBr, NH3, NO2, HCN, PCl3, ClO2, BF3, HNO3, F2, Br2, I2 and Cl2 (Bacon et al. 1991); this is important for the detection of dangerous leakages in industrial factories.
Ion mobility spectrometry is an analytical technique with a promising future due to increased terrorism and drug trafficking since it is the technique of choice for rapid and low cost detection of illicit drugs and explosives. IMS portability and easy operation make it an essential tool for military, police and security personnel.
Conflicts of interest: The manuscript was prepared and revised by the author, who declares the absence of any conflict which can put the validity of the presented review in risk.
BIBLIOGRAPHY
1. ALBRITTON, D.L.; MILLER, T.M.; MARTIN, D.W.; MCDANIEL, E.W. 1968. Mobilities of mass-identified H3+ and H+ ions in hydrogen. Phys. Rev. 171(1):94-102. [ Links ]
2. ASBURY, G.R.; KLASMEIER, J.; HILL, Jr. H.H. 2000. Analysis of explosives using electrospray ionization/ ion mobility spectrometry (ESI/IMS). Talanta. 50(6):1291-1298. [ Links ]
3. ASBURY, G.R.; HILL, Jr. H.H. 2000. Using different drift gases to change separation factors (a) in ion mobility spectrometry. Anal. Chem. 72(3):580-584. [ Links ]
4. BACON, A.T.; GETZ, R.; REATEGUI, J. 1991. Ion-mobility spectrometry tackles tough process monitoring. J. Chem. Eng. Program. 87(6):61-64. [ Links ]
5. BAIM, M.A.; HILL, Jr. H.H. 1982. Tunable selective detection for capillary gas chromatography by ion mobility monitoring. Anal. Chem. 54(1):38-43. [ Links ]
6. BAIM, M.A.; EATHERTON, R.L.; HILL, Jr H.H. 1983. Ion mobility detector for gas chromatography with a direct photoionization source. Anal. Chem. 55(11):1761-1766. [ Links ]
7. BERANT, Z.; KARPAS, Z. 1989. Mass-mobility correlation of ions in view of new mobility data. J. Am. Chem. Soc. 111(11):3819-3824. [ Links ]
8. BESSA, V.; DARWICHE, K.; TESCHLER, H.; SOMMERWERCK, U.; RABIS, T.; BAUMBACH, J.I.; FREITAG, L. 2011. Detection of volatile organic compounds (VOCs) in exhaled breath of patients with chronic obstructive pulmonary disease (COPD) by ion mobility spectrometry. Int. J. Ion Mob. Spectrom. 14(1):7-13. [ Links ]
9. BLUHM, B.K.; GILLIG, K.J.; RUSSELL, D.H. 2000. Development of a Fourier-transform ion cyclotron resonance mass spectrometer-ion mobility spectrometer. Rev. Sci. Instrum. 71(11):4078-4086. [ Links ]
10. BOCOS-BINTINTAN, V.; MOLL, V.H.; FLANAGAN, R.J.; THOMAS, C.L.P. 2010. Rapid determination of alcohols in human saliva by gas chromatography differential mobility spectrometry following selective membrane extraction. Int. J. Ion Mob. Spectrom. 13(2):55-63. [ Links ]
11. BOGGIO, N.G.; PIERPAULI, K.; RINALDI, C.A.; LAMAGNA, A. 2011. Detection of volatile organic compound with an ion mobility spectrometry cell type device with a corona discharge ionization source. Sensor Lett. 9(2):866-868. [ Links ]
12. BOLLAN, H.R.; STONE, J.A.; BROKENSHIRE, J.L.; RODRIGUEZ, M.R.; EICEMAN, G.A. 2007. Mobility resolution and mass analysis of ions from ammonia and hydrazine complexes with ketones formed in air at ambient pressure. J. Am. Soc. Mass Spectrom. 18(5):940-951. [ Links ]
13. BOTA, G.M.; HARRINGTON, P.B. 2006. Direct detection of trimethylamine in meat food products using ion mobility spectrometry. Talanta 68(3):629-635. [ Links ]
14. BUNKOWSKI, A.; MADDULA, S.; DAVIES, A.N.; WESTHOFF, M.;LITTERST, P.; BÖDEKER, B.; BAUMBACH, J.I. 2010. One year time series of investigations of analytes within human breath using ion mobility spectrometry. Int. J. Ion Mobil. Spectrom. 13(3-4):141-148. [ Links ]
15. CARSTENS, E.T.H.; HIRN, A.; QUINTEL, M.; NOLTE, J.; JÜNGER, M.; PERL, T.; VAUTZ, W. 2010. Online determination of serum propofol concentrations by expired air analysis. Int. J. Ion Mob. Spectrom. 13(1):37-40. [ Links ]
16. CLOWERS, B.H.; HILL, H.H. 2005. Mass analysis of mobility-selected ion populations using dual gate, ion mobility, quadrupole ion trap mass spectrometry. Anal. Chem. 77(18):5877-5885. [ Links ]
17. COHEN, M.J.; KARASEK, F.W. 1970. Plasma chromatography - a new dimension for gas chromatography and mass spectrometry. J. Chromatogr. Sci. 8(6):330-337. [ Links ]
18. DE HOFFMANN, E.; STROOBANT, V. 2001. Mass spectrometry: principles and applications. 3rd edition. John Wiley and Sons. 493p. [ Links ]
19. DWIVEDI, P.; WU, C.; MATZ, L.M.; CLOWERS, B.H.; SIEMS, W.F.; HILL, Jr. H.H. 2006. Chiral separation by ion mobility spectrometry. Anal. Chem. 78(24):8200- 8206. [ Links ]
20. DWIVEDI, P.; PUZON, G.; TAM, M.; LANGLAIS, D.; JACKSON, S.; KAPLAN, K.; SIEMS, W.F.; HILL, H.H. 2010. Metabolic profiling of Escherichia coli by ion mobility-mass spectrometry with MALDI ion source. J. Mass Spectrom. 45(12):1383-1393. [ Links ]
21. DWORZANSKI, J.P.; TRIPATHI, A.; SNYDER, A.P.; MASWDEH, W.M.; WICK, C.H. 2005. Novel biomarkers for Gram-type differentiation of bacteria by pyrolysis-gas chromatography-mass spectrometry. J. Anal. Appl. Pyrolysis 73(1):29-38. [ Links ]
22. EATHERTON, R.L.; MORRISSEY, M.A.; HILL, H.H. 1988. Comparison of ion mobility constants of selected drugs after capillary gas chromatography and capillary supercritical fluid chromatography. Anal. Chem. 60(20):2240-2243. [ Links ]
23. EICEMAN, G.A.; SALAZAR, M.R.; RODRIGUEZ, M.R.; LIMERO, T.F.; BECK, S.W.; CROSS, J.H.; YOUNG, R.; JAMES, J.T. 1993. Ion mobility spectrometry of hydrazine, monomethylhydrazine, and ammonia in air with 5-nonanone reagent gas. Anal. Chem. 65(13):1696-1702. [ Links ]
24. EICEMAN, G.A.; WANG, Y.F.; GARCIA-GONZALEZ, L.; HARDEN, C.S.; SHOFF, D.B. 1995. Enhanced selectivity in ion mobility spectrometry analysis of complex mixtures by alternate reagent gas chemistry. Anal. Chim. Acta 306(1):21-33. [ Links ]
25. EICEMAN, G.A.; NAZAROV, E.G.; STONE, J.A. 2003. Chemical standards in ion mobility spectrometry. Anal. Chim. Acta 493(2):185-194. [ Links ]
26. EICEMAN, G.A.; KARPAS, Z. 2005. Ion mobility spectrometry. 2nd edition. Boca Raton: FL.CRC Press. 350p. [ Links ]
27. ELLIS, H.W.; PAI, R.Y.; GATLAND, I.R.; MCDANIEL, E.W.; WERNLUND, R.; COHEN, M.J. 1976. Ion identity and transport properties in CO2 over a wide pressure range. J. Chem. Phys. 64(10):3935-3941. [ Links ]
28. EXCELLIMS CORPORATION. 2011. Available in internet in: http://www.excellims.com Accessed on 12/2/2011. [ Links ]
29. FERNÁNDEZ-MAESTRE, R.; HILL Jr., H.H. 2009. Ion mobility spectrometry for the rapid analysis of over- the-counter drugs and beverages. Int. J. Ion Mob. Spectrom. 12(1):3-14. [ Links ]
30. FERNÁNDEZ-MAESTRE, R.; HARDEN, C.S.; EWING, R.G.; CRAWFORD, C.L.; HILL Jr., H.H. 2010a. Chemical standards in ion mobility spectrometry. Analyst 135(6):1433-1442. [ Links ]
31. FERNÁNDEZ-MAESTRE, R.; WU, C.; HILL, H.H. 2010b. Using a buffer gas modifier to change separation selectivity in ion mobility spectrometry. Int. J. Mass Spectrom. 298(1-3):2-9. [ Links ]
32. FERNÁNDEZ-MAESTRE, R.; WU, C.; HILL Jr., H.H. 2012. Buffer gas modifiers effect resolution in ion mobility spectrometry through selective ion-molecule clustering reactions. Rap. Comm. Mass Spectrom. Accepted. [ Links ]
33. FINTHAMMER, M.; BEIERLE, C.; FISSELER, J.; KERN-ISBERNER, G.; BAUMBACH, J.I. 2010. Using probabilistic relational learning to support bronchial carcinoma diagnosis based on ion mobility spectrometry. Int. J. Ion Mob. Spectrom. 13(2):83- 93. [ Links ]
34. GAN, T.H.; CORINO, G.T. 2000. Selective detection of alkanolamine vapors by ion mobility spectrometry with ketone reagent gases. Anal. Chem. 72(4):807- 815. [ Links ]
35. GUHARAY, S.K.; DWIVEDI, P.; HILL, H.H. 2008. Ion mobility spectrometry: ion source development and applications in physical and biological sciences. IEEE T. Plasma Sci. 36(4):1458-1470. [ Links ]
36. GUNZER, F.; ULRICH, A.; BAETHER, W. 2010. A novel non-radioactive electron source for ion mobility spectrometry. Int. J. Ion Mob. Spectrom. 13(1):9-16. [ Links ]
37. HALLEN, R.W.; SHUMATE, C.B.; SIEMS, W.F.; TSUDA, T.; HILL, H.H. 1989. Preliminary investigation of ion mobility spectrometry after capillary electrophoretic introduction. J. Chromatogr. A. 480:233-245. [ Links ]
38. HILL, Jr., H.H.; SIMPSON, G. 1997. Capabilities and limitations of ion mobility spectrometry for field screening applications. J. Field Anal. Chem. Technol. 1(3):119-134. [ Links ]
39. JOHNSON, P.V.; BEEGLE, L.W.; KIM, H.I.; EICEMAN, G.A.; KANIK, I. 2007. Ion mobility spectrometry in space exploration. Int. J. Mass Spectrom. 262(1- 2):1-15. [ Links ]
40. KANU, A.B.; HILL Jr., H.H. 2008. Ion mobility spectrometry detection for gas chromatography. J. Chromatogr. A. 1177(1):12-27. [ Links ]
41. KANU, A.B.; DWIVEDI, P.; TAM, M.; MATZ, L.; HILL, H.H. Jr. 2008. Ion mobility-mass spectrometry. J. Mass Spectrom. 43(1):1-22. [ Links ]
42. KARASEK, F.W 1970. Drift-mass spectrometer. Res. Develop. 21(12):25 (cited in KARASEK F.W 1972. Trace analysis and fundamental studies by plasma chromatography. Int. J. Environ. Anal. Chem. 21(12):157-166. [ Links ]
43. KIM, S.H.; KARASEK, F.W.; ROKUSHIKA, S. 1978. Plasma chromatography with ammonium reactant ions. Anal. Chem. 50(1):152-155. [ Links ]
44. KOJIRO, D.R.; COHEN, M.J.; STIMAC, R.M.; WERNLUND, R.F.; HUMPHRY, D.E.; NORISHIGE, T. 1991. Determination of C1-C4 alkanes by ion mobility spectrometry. Anal. Chem. 63(20):2295- 2300. [ Links ]
45. LANGEVIN, P. 1903. L'ionization des gaz. Ann. Chim. Phys. 28:289-384. [ Links ]
46. LAWRENCE, A.H.; NEUDORFL, P. 1988. Detection of ethylene glycol dinitrate vapors by ion mobility spectrometry using chloride reagent ions. Anal. Chem. 60(2):104-109. [ Links ]
47. LAWRENCE, A.H.; BARBOUR, R.J.; SUTCLIFFE, R. 1991. Identification of wood species by ion mobility spectrometry. Anal. Chem. 63(13):1217-1221. [ Links ]
48. LEE, D-S.; WU, C.; HILL Jr., H.H. 1998. Detection of carbohydrates by electrospray ionization - ion mobility spectrometry following microbore high- performance liquid chromatography. J. Chromatogr. A. 822(1):1-9. [ Links ]
49. LOVELOCK, J.E.; WASILEWSKA, E.M. 1949. An ionisation anemometer. J. Sci. Instr. 26(11):367-370. [ Links ]
50. LUBMAN, D.M.; KRONICK, M.N. 1982a. Plasma chromatography with laser-produced ions. Anal. Chem. 54(9):1546-1551. [ Links ]
51. LUBMAN, D.M.; KRONICK, M.N. 1982b. Discrimination of isomers of xylene by resonance enhanced two- photon ionization. Anal. Chem. 54(13):2289-2291. [ Links ]
52. MÁRQUEZ-SILLERO, I.; AGUILERA-HERRADOR, E.; CÁRDENAS; S.; VALCÁRCEL, M. 2011.Ion-mobility spectrometry for environmental analysis. TrAC Trends Anal. Chem. 30(5):677. [ Links ]
53. MASON, E.A.; SCHAMP, Jr. H.W. 1958. Mobility of gaseous ions in weak electric fields. Ann. Phys. 4(3):233-270. [ Links ]
54. McDANIEL, E.W.; MARTIN, D.W.; BARNES, W.S. 1962. Drift-tube mass spectrometer for studies of low- energy ion-molecule reactions. Rev. Sci. Instr. 33(1):2-7. [ Links ]
55. McDANIEL, E.W. 1964. Collisional phenomena in ionized gases, New York: Wiley. 242p. [ Links ]
56. McLEAN, J.A.; RUOTOLO, B.T.; GILLIG, K.J.; RUSSELL, D.H. 2005. Ion mobility-mass spectrometry: a new paradigm for proteomics. Int. J. Mass Spectrom. 240(3):301-315. [ Links ]
57. McMINN, D.G.; KINZER, J.A.; SHUMATE, C.B.; SIEMS, W.F.; HILL Jr., H.H. 1990. Ion mobility detection following liquid chromatographic separation. J. Microcolumn Sep. 2(4):188-192. [ Links ]
58. MENG, Q.; KARPAS, Z.; EICEMAN, G.A. 1995. Monitoring indoor ambient atmospheres for volatile organic compounds using an ion mobility analyzer array with selective chemical ionization. Int. J. Environ. Anal. Chem. 61(2):81-94. [ Links ]
59. MORRISSEY, M.A.; WIDMER, H.M. 1991. Ion-mobility spectrometry as a detection method for packed- column supercritical fluid chromatography. J. Chromatogr. 552:551-561. [ Links ]
60. MYUNG, S.; WISEMAN, J.M.; VALENTINE, S.J.; TAKATS, Z.; COOKS, R.G.; CLEMMER, D.E. 2006. Coupling desorption electrospray ionization with ion mobility / mass spectrometry for analysis of protein structure: evidence for desorption of folded and denatured states. J. Phys. Chem. B 10 (10):5045-5051. [ Links ]
61. PROCTOR; C.J.; TODD, J.F.J. 1984. Alternative reagent ions for plasma chromatography. Anal. Chem. 56(11):1794-1797. [ Links ]
62. PUTON, J.; NOUSIAINEN, M.; SILLANPAA, M. 2008. Ion mobility spectrometers with doped gases. Talanta 76(5):978-987. [ Links ]
63. REARDEN, P.; HARRINGTON, P.B. 2005. Rapid screening of precursor and degradation products of chemical warfare agents in soil by solid-phase microextraction ion mobility spectrometry (SPME-IMS). Anal. Chim. Acta 545(1):13-20. [ Links ]
64. REVERCOMB, H.E.; MASON, E.A. 1975. Theory of plasma chromatography/ gaseous electrophoresis. Anal. Chem. Rev. 47(7):970-983. [ Links ] [ Links ]
66. RUTHERFORD, E. 1897. The velocity and rate of recombination of the ions of gases exposed to Rontgen radiation. Phil. Mag. 44(270):422-440. [ Links ]
67. RUTHERFORD, E. 1899. Uranium radiation and the electrical conduction produced by it. Phil. Mag. 47(284):109-163. [ Links ]
68. SABO, M.; MATEJCIK, S. 2011. Ion mobility spectrometry for monitoring high-purity oxygen. Anal. Chem. 83(6):1985-1989. [ Links ]
69. SHEIBANIA, A.; TABRIZCHI, M.; GHAZIASKAR, H.S. 2011. Determination of methadone in human hair by headspace extraction and ion mobility spectrometry. Anal. Lett. 44(4):667-675. [ Links ]
70. SHUMATE, C.B.; ST. LOUIS, R.H.; HILL Jr., H.H. 1986. Table of reduced mobility values from ambient pressure ion mobility spectrometry. J. Chromatogr. 373:141-173. [ Links ]
71. SHUPP, A.M.; RODIER, D.; ROWLEY, S. 2007. Monitoring airborne molecular contamination: a quantitative and qualitative comparison of real-time and grab- sampling techniques. Proc. SPIE, the International Society for Optical Engineering 6518, 65183Z. doi:10.1117/12.707776. [ Links ]
72. SNYDER, A.P.; SHOFF, D.B.; EICEMAN, G.A.; BLYTH, D.A.; PARSONS, J.A. 1991. Detection of bacteria by ion mobility spectrometry. Anal. Chem. 63(5):526- 529. [ Links ]
73. SPANGLER, G.E.; CARRICO, J.P. 1983. Membrane inlet for ion mobility spectrometry. Int. J. Mass Spectrom. Ion Phys. 52(2-3):267-287. [ Links ]
74. SPANGLER, G.E.; CAMPBELL, D.N.; CARRICO, J.P. 1985. Recent Advances in ion mobility spectrometry for explosives vapor detection. J. Test Eval. 13(3):234- 241. [ Links ]
75. ST. LOUIS, R.H.; SIEMS, W.F.; HILL Jr., H.H. 1989. Evaluation of direct axial sample introduction for ion mobility detection after capillary gas chromatography. J. Chromatogr. 479(2):221-231. [ Links ]
76. ST. LOUIS, R.H.; HILL, Jr.; H.H.; EICEMAN, G.A. 1990. Ion mobility spectrometry in analytical chemistry. CRC Cr. Rev. Anal. Chem. 21(5):321-355. [ Links ]
77. TABRIZCHI, M.; ROUHOLAHNEJAD, F. 2004. Corona discharge ion mobility spectrometry at reduced pressures. Rev. Sci. Instr. 75(11):4656-4662. [ Links ]
78. TIEBE, C.; HÜBERT, T.; KOCH, B.; RITTER, U.; STEPHAN, I. 2010. Investigation of gaseous metabolites from moulds by ion mobility spectrometry (IMS) and gas chromatography-mass spectrometry (GC-MS). Int. J. Ion Mob. Spectrom. 13(1):17-24. [ Links ]
79. UGAROV, M.V.; EGAN, T.; KHABASHESKU, D.V.; SCHULTZ, J.A.; PENG, H.; KHABASHESKU, V.N.; FURUTANI, H.; PRATHER, K.S.; WANG, J.H.W.; JACKSON, S.N.; WOODS, A.S. 2004. MALDI matrices for biomolecular analysis based on functionalized carbon nanomaterials. Anal. Chem. 76(22):6734-6742. [ Links ]
80. VALENTINE, S.J.; COUNTERMAN, A.E.; HOAGLUND, C.S.; REILLY, J.P.; CLEMMER, D.E. 1998. Gas-phase separations of protease digests. J. Am. Soc. Mass Spectrom. 9(11):1213-1216. [ Links ]
81. VAUTZ, W.; BAUMBACH, J.I.; UHDE, E. 2006. Detection of emissions from surfaces using ion mobility spectrometry. Anal. BioAnal. Chem. 384(4):980-986. [ Links ]
82. WEIS, P. 2005. Structure determination of gaseous metal and semi-metal cluster ions by ion mobility spectrometry. Int. J. Mass Spectrom. 245(1-3):1-13. [ Links ]
83. WU, C.; SIEMS, W.F.; HILL Jr., H.H.; HANNAN, R.M. 1998. Analytical determination of nicotine in tobacco by supercritical fluid chromatography-ion mobility detection. J. Chromatogr. A. 811(1-2):157-161. [ Links ]
84. WU, C.; SIEMS, W.F.; HILL Jr., H.H. 2000. Secondary electrospray ionization ion mobility spectrometry/ mass spectrometry of illicit drugs. Anal. Chem. 72(2):396-403. [ Links ]
85. WYTTENBACH T.; VON HELDEN G.; BOWERS M.T. 1996. Gas-phase conformation of biological molecules:â bradykinin. J. Am. Chem. Soc. 118(35):8355-8364 [ Links ]
86. YUAN, A.; ALIAGA-ROSSEL, R.; CHOI, P.; GILLES, J.P. 2005. Development of a short pulsed corona discharge ionization source for ion mobility spectrometry. Rev. Sci. Instr. 76(8):085105-085105-6. [ Links ]
Recibido: Septiembre 26 de 2011 Aceptado: Julio 19 de 2012
