











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCTION
Enzootic bovine leukemia (EBL) is a disease that primarily affects dairy cattle. Infected cattle show no clinical symptoms, but in most cases, they develop persistent lymphocytosis due to proliferation of B cells; in others, the infected bovine present the tumor form or lymphosarcomas (Aida et al. 2013). This disease causes losses in yield and reproductive levels in infected cattle; studies have shown that herds infected with BLV had a decrease in milk production from 2,5 to 28,14% compared to the production of the uninfected herd (Emanuelsson et al. 1992; Nekouei et al. 2016) and increased cull rate, as well as susceptibility to other infectious diseases as mastitis (Kakinuma et al. 2014), diarrhea and pneumonia (Emanuelsson et al. 1992). The etiological agent of EBL is the bovine leukemia virus (BLV), a retrovirus belonging to the Deltaretrovirus genus (Wu et al. 2003). BLV is related, from structural and functional point of view, with human T-cell lymphotropic viruses (HTLV-1, HTLV-2 and HTLV-3) and simian T-cell lymphotropic virus (STLV-1) (Hulo et al. 2011). BLV has become a very important tool for HTLV-1 study (Gillet et al. 2007; Aida et al. 2013), since the infection produced by both viruses is similar, affecting either T-cells (HTLV) or B-cells (BLV), none of them produce detectable viremia and their genomic organization is the same. BLV genome is a single stranded RNA of positive sense, consists of three genes: gag, pol and env, encoding different proteins: p15, p12 and p24 proteins are codified by gag gene; integrase enzyme (IN) and reverse transcriptase (RT) enzyme are codified by pol gene; gp51 and gp30 glycoproteins are codified by env gene (Gillet et al. 2007); moreover, BLV has a pX region that codifies for different regulatory and incidental proteins (tax, rex, G4, G3). Among them viral transcription activators (tax), viral mRNA exporters, and structural protein expression (rex), high viral load maintenance proteins (G4), and low viral load proteins (R3) (Gillet et al. 2007). Env gene is highly conserved and the antigen gp51 is generally present in all animals during different stages of infection (Baruta et al. 2011). Variation in env gene sequence have allowed to identify nine genotypes of BLV (Polat et al. 2016). The most prevalent genotypes worldwide are 1, 2 and 3 (Inoue et al. 2011). In America BLV is widely distributed and these genotypes have been found: in the United States, 1 and 2; in Costa Rica, 1 and 5 (Zhao el al. 2007); in Brazil, 1, 2 and 6 (Camargos et al. 2002); in Chile, 7 (Felmer et al. 2005); in Uruguay, 1 (Moratorio et al. 2010), and in Bolivia, 9 (Polat et al. 2016). The aim of this study was to identify the genotypes of BLV in different municipalities of Antioquia, Colombia.
MATERIALS AND METHODS
Animals and samples: This research is descriptive and only the population sample was evaluated in a moment of time. The sequenced amplicons were selected according to the municipality of origin. This investigation was of exploratory type and therefore it tries to give a first approximation to the possible genotypes that circulate in the dairy herds of the provice of Antioquia. Blood samples were drawn from 500 Holstein cows, which were located in herds of six municipalities of Antioquia: Bello, Belmira, Entrerríos, Medellín, Rionegro and San Pedro de los Milagros. Sampling was approved by the ethics committee of the National University of Colombia (CEMED-022, July 13, 2015).
DNA extraction: Sampling was done with an 18G needle with vacuum vacutainer system (DBvacutainer®) and EDTA as an anticoagulant; samples were homogenized by inversion and transferred to the laboratory under refrigeration for DNA extraction. DNA from samples was obtained by the salting out technique (Miller et al. 1988), re-suspended in TE 1X pH 8.0 (Tris HCl 1M and EDTA 0.5M) and stored at 4°C until analysis. The quality and quantity of DNA obtained was evaluated in a spectrophotometer (NanoDrop 2000®, Massachusetts-USA) and agarose gel 1%.
Nested PCR: A region of the viral env gene (gp51) was amplified to obtain a 444bp fragment with the primers reported by Beier et al. (2001). The first round of PCR was performed in a final volume of 25µl with 150ng DNA, 3.0µl of 10mM of each primer env 5023 (5´-TCTGTGCCAAGTCTCCCAGATA-3´) and env 5608 (5´-AACAACAACCTCTGGGGAGGGT-3´), 0.4mM dNTPs, 1X buffer PCR (ThermoScientific®), 3mM MgCl2 and 1U Taq Polymerase DNA. In the second PCR reaction 5µl of the first amplification PCR product was used as template DNA, with same concentrations of other reagents and env primers 5099 (5´-CCCACAAGGGCGGCGCCGGTTT-3´) and env 5521 (5´-GCGAGGCCGGGTCCAGAGCTGG-3´) in a final volume of 30µL. Reactions for both PCR were identical, and were performed in a T3 thermocycler (Biometra®) with the following protocol: initial denaturation at 94°C for 5 minutes, followed by 40 cycles of 94°C for 30 seconds, 60°C for 30 seconds, and 72°C for 1 minute; the final extension was run at 72°C for 5 minutes. As a negative and positive control, we run PCR reactions without template DNA and with DNA of a cow that tested positive in a previous study (Úsuga-Monroy, et al. 2015), respectively. The product of the second reaction was checked on an agarose gel stained with 2% EZ-VISION (Amresco®) in a Gel Doc (BioRad, California-USA).
Sequencing and phylogenetic analysis: Eight amplicons sent for sequencying in a commercial facility (Macrogen Inc. Korea); these amplicons were from different herds located at these municipalities in the province of Antioquia: Bello (1), Belmira (2), Entrerríos (1), Medellín (2) Rionegro (1), and San Pedro de Los Milagros (1). The nucleotide sequences of the samples were compared with 53 partial sequences of the viral env gene registered in GenBank representative of the 9 BLV genotypes and representing different geographic regions including several South American countries. Sequences were manually aligned in the software MEGA V7. Phylogenetic analysis was performed using two methods. First, a Maximum Likelihood analysis using MEGA V7 was performed. The Kimura 2-parameters with Gamma distribution (K2+G) was best substitution model based on the Bayesian Information Criterion (BIC). Bootstrap values were determined with 1000 repetitions and only significant values over 70% were considered as significant. A second phylogenetic tree was built by Bayesian methods using MrBayes V3.2.2. The evolution model used was Kimura 2-parameters with Gamma (K2+G) distribution. Convergence (average standard deviation of split sequences) of 0.005 was obtained after 1.000.000 generations. Trees were sampled every 1000 generations. The consensus tree was obtained after discarding (burn in) the initial 25% of the generations. The final tree was edited in FigTree V1.4.0.
RESULTS AND DISCUSSION
PCR is highly sensitive methodology of direct diagnosis for infection with BLV (Rola et al. 2002). PCR helps to rationally direct diagnostic tests for early diagnosis and disease control plans. The BLV is widely distributed in the world, probably migrated from Europe to America through infected cattle. It is considered that in America the disease is enzootic. Molecular prevalence for bovine leukemia virus in this study was 44% of infection, other South American countries show lower molecular prevalences as for example Chile (27,9%), Venezuela (33,3%), Bolivia (30,7%) and Peru (42,3%); however, countries such as Paraguay (54,7%), Brazil (60,8%) and Argentina (77,4%) have higher molecular prevalences (Polat et al. 2017).
The most studied BLV gene is the envelope gene, which translates for the gp51 glycoprotein. The gp51 has an essential role in the life cycle of the virus, is required to enter the cell and as a target of neutralizing antibodies. The BLV clusters have been carried out through the phylogenetic analysis of a 444pb fragment of the envelope gene (env) of the virus (Beier et al. 2001), these studies allow to identify the distribution of BLV in the region and to reconstruct its evolutionary history. In addition, small differences have been demonstrated in many ways for the gp51 mutations which are related to the geographic location of the simple, allowing to group the BLV in ten genotypes (Polat et al. 2016). Proviral BLV DNA was detected in 219 samples and 281 specimens tested negative by nested PCR. Eight positive PCR products were selected for sequencing. Their sequences were aligned to 53 sequences of the env gene from the following countries: United States, Costa Rica (North and Central America), Argentina, Brazil, Uruguay, Paraguay, Peru, Bolivia, Chile (South America), Germany, Russia, Belgium, Poland, Ukraine, Croatia (Europe), Japan, (Asia) and Australia (Oceania). In the Maximum Likelihood phylogenetic the sequences from Bello, Rionegro, San Pedro, Entrerríos, Medellín and one sequence from Belmira were grouped to isolates from United States, Argentina and Japan in genotype 1, with a branch support of 79%. One out of the eight sequences analyzed from the Belmira was cluster within genotype 3, with sequences from United States and Japan; the branch support for this clade was 97%. Thus, the municipality of Belmira showed both genotypes, 1 and 3 (Figure 1). A phylogenetic analysis was also performed by Bayesian methods in which 7 out of the 8 sequences analyzed were also clustered in genotype 1, and only one sequence from the municipality of Belmira was grouped into genotype 3; branch support values, expressed in posterior probabilities, were 100% in both clades (Figure 2). The results of both phylogenetic reconstructions obtained by Maximum Likelihood and Bayesian methods were similar.
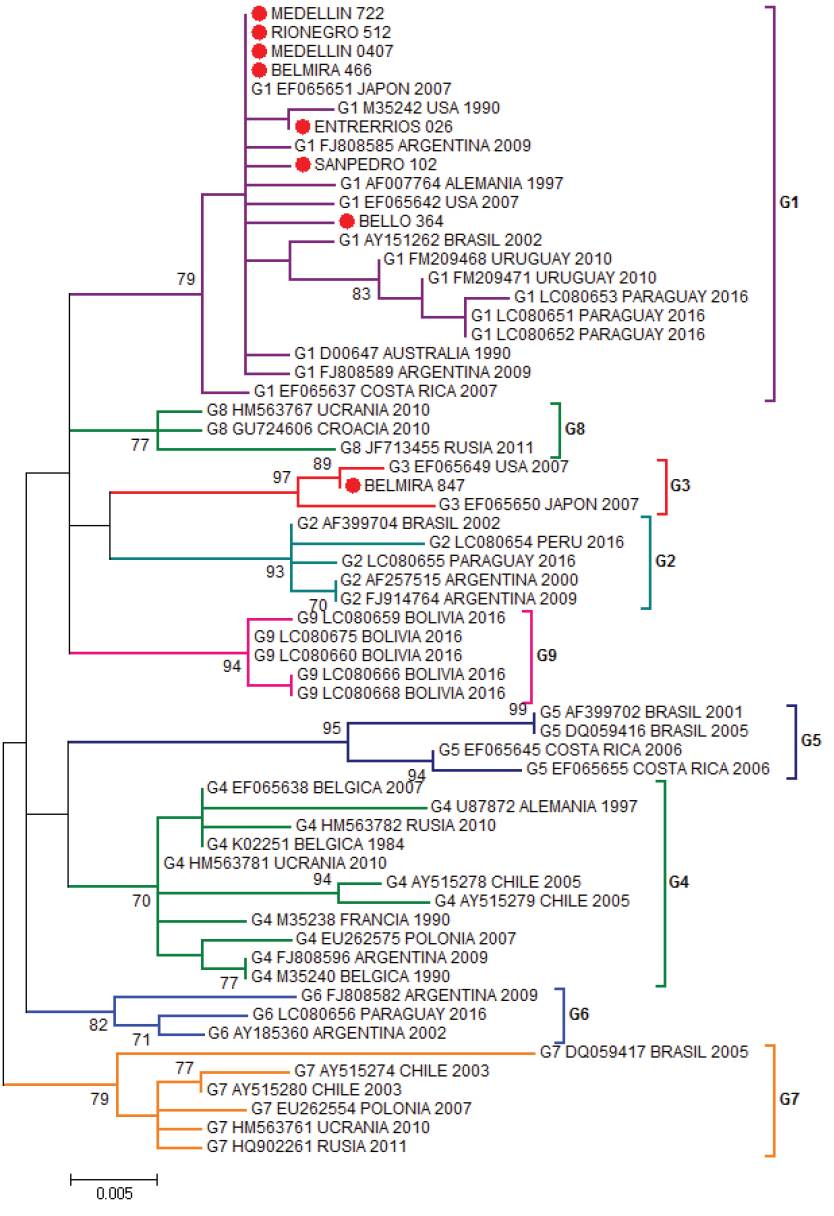
Figure 1 Phylogenetic tree constructed by Maximum Likelihood using as substitution model Kimura 2-parameters with Gamma distribution (K2+G). Boostrap values are indicated as data from 1000 replicas (Bar 0.005 substitutions per site) based on partial sequences of 400 nucleotides of BLV env gene. Colombian sequences are indicated as (●) and were compared with 53 reported in GenBank sequences for the env gene. Genotype, access numbers and country of origin of the sequences indicated.
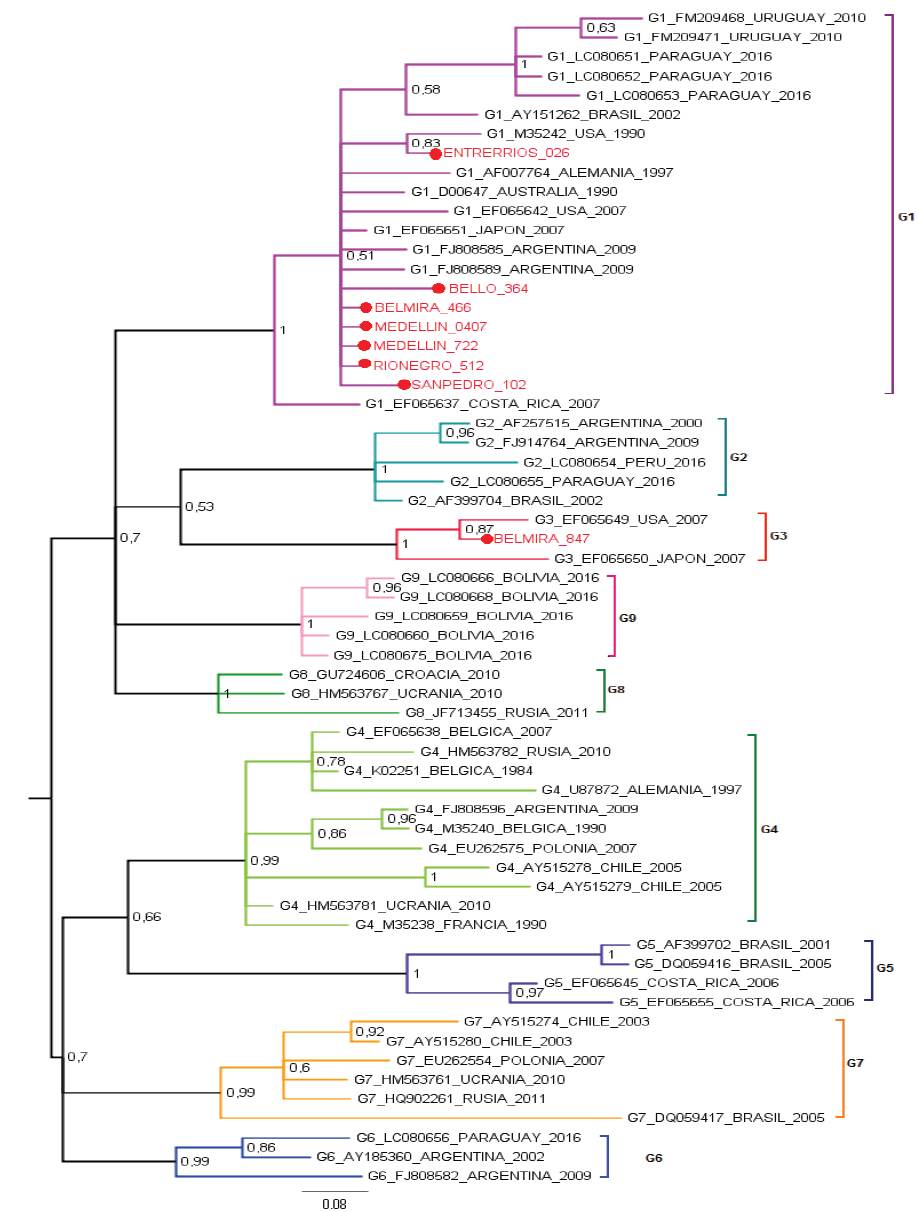
Figure 2 Bayesian phylogenetic tree using Kimura 2-parameters with Gamma distribution (K2+G). Posterior probability values are indicated as numbers on the bar. The analysis is based on partial sequences of 400 nucleotides of BLV env gene. Colombian sequences are indicated as (●) and were compared with 53 reported in GenBank for env gene. Genotype, access numbers and country of origin of the sequences indicated.
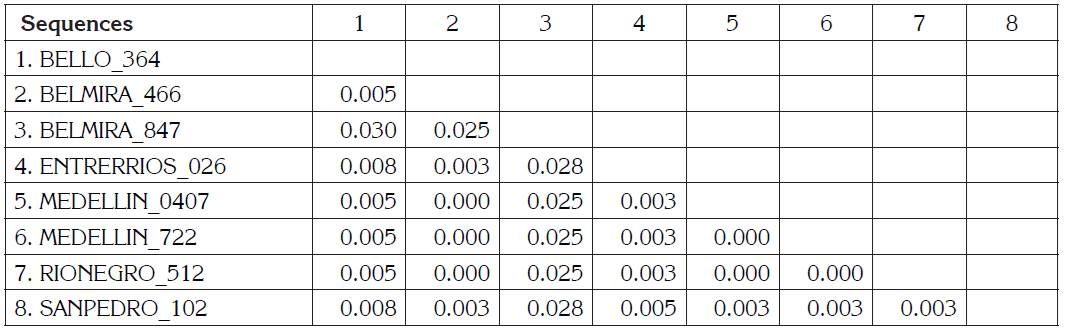
BLV sequences BELMIRA_466, MEDELLÍN_0407, MEDELLIN_722, RIONEGRO_512 are the identical using p-distance model, although they are geographically remote municipalities. BELMIRA_847 has a higher proportion of nucleotide sites different than other samples and is farther from BELLO_364 sequence (Table 1). ENTRERRIOS_026 sequence formed a clade with M35242 sequence from the United States, with a posterior probability value of 83% for Bayesian phylogenetic tree. BELMIRA _847 sequence was the only one grouped in genotype 3 along with sequences from the United States and Japan with a posterior probability value of 100% for Bayesian phylogenetic tree (Figure 2) and branch supporting value of 97% for the phylogenetic tree by Maximum Likelihood (Figure 1).
Table 1 Estimates of Evolutionary Divergence between Sequences. The number of base differences per site from between sequences are shown. The analysis involved 8 nucleotide sequences. Codon positions included were 1st+2nd+3rd+Noncoding. All positions with less than 95% site coverage were eliminated. That is, fewer than 5% alignment gaps, missing data, and ambiguous bases were allowed at any position. There were a total of 400 positions in the final dataset. Evolutionary analyses were conducted in MEGA V7.
Maximum Likelihood analysis and Bayesian analysis grouped one sample from the municipality of Belmira in genotype 3, while the remaining 7 samples were grouped in genotype 1. The municipality of Belmira was the only one presenting both genotypes 1 and 3 in samples from the same municipality. The presence of the two genotypes (1 and 3) in the municipality of Belmira indicates that the BLV has probably entered the country through the purchase of livestock and import of artificial insemination straws on several occasions, other studies have also reported the presence of two or more genotypes in one herd (Ababneh et al. 2012; Mekata et al. 2015). Genotypes 1 and 3 are circulating in specialized dairy farms in Antioquia, since there is no limitation for viral transmission. Recently the viral load has been linked to horizontal transmission because a cow infected with less than 3 copies of the virus per 100 cells cannot spread BLV to other cattle of more than 30 months old (Mekata et al. 2015). Transmission of the virus can be vertical and horizontal. In vertical transmission colostrum is a vehicle for the virus (Nagy et al. 2007), also exosomes in milk from infected cows contain BLV proteins, they are transmitted by an alternative route different from the viral infection and have an important role in removing proteins through BLV infected cells (Yamada et al. 2013). In the horizontal transmission reusing needles for vaccination and reuse of gloves during palpation or dehorning (Gillet et al. 2007; Ortega et al. 2016) help to spread the virus. Infection through semen is also included in horizontal transmission (Dus Santos et al. 2007).
The most prevalent genotypes BLV worldwide are 1, 2 and 3 (Inoue et al. 2011). In America, the following genotypes are found: genotypes 1, 3 and 4 are found in the United States, 1 and 5 are in Costa Rica (Zhao et al. 2007), 1, 2, 5, 6 and 7 are in Brazil (Camargos et al. 2002; Moratorio et al. 2010), 4 and 7 in Chile (Felmer et al. 2005), 1 in Uruguay (Moratorio et al. 2010), 1, 2, 4 and 6 in Argentina (Rodriguez et al. 2009), 1, 2 and 6 un Perú (Polat et al. 2016), 1, 2 and 6 in Paraguay (Polat et al. 2016) and 1, 2, 6 and 9 in Bolivia (Polat et al. 2016). Genotypes 1 and 2 were reported in dairy cattle in the province of Nariño, Colombia (Benavides et al. 2017), while in our study in the province of Antioquia, genotypes 1 and 3 were found in Holstein cows. According to Inoue et al. (2011) the most frequent BLV genotypes are 1, 2 and 3. These data suggest that genotype 1 is the most widespread, it is also the most frequent detected in samples from Antioquia, Colombia in this research. For now, no relationship has been reported between genotype and virulence or resistance, this may be due to the few changes at the nucleotide level that the virus presents. The low mutation rate of BLV is due to the long generation time of Deltaretrovirus. BLV mutation rate is 1.7x10-5 s/n/c is similar to the human T-cell lymphotropic virus (HTLV-1) 1.6x10-5 s/n/c mutation rate (Sanjuán et al. 2010). Bovine leukemia virus genotypes 1 and 3 are circulating in dairy herds in the province of Antioquia, Colombia. The two genotypes can be found in the same municipality. It is important to maintain good iatrogenic practices such as: do not reuse needles, palpation gloves, do not use colostrum or milk from infected mothers in healthy calves. In this way, the dispersion of viral particles within and between herds can be controlled. On the other hand, it is important to evaluate more dairy herds and other fragments of the viral genome to establish genetic variability analysis for BLV and to have a better focus about the distribution of the virus in the dairy herds of the department and the country.

