











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCTION
Gastric cancer is fifth in occurrence worldwide after breast, lung, colorectal, and prostate cancers (http://globocan.iarc.fr/Default.aspx). In 2012, there were 951,594 new cases of gastric cancer reported in the world, which represent 6.8% of the total number of cancer cases; in the Colombian population, we observe an incidence rate of 13.6 cases for every 100.000 inhabitants (http://globocan.iarc.fr/Default.aspx). In this same year, 723.073 people with gastric cancer died, a number that represents 8,8% of deaths caused by cancer; the mortality rate in Colombia for that year was 11.3 for every 100.000 inhabitants (http://globocan.iarc.fr/Default.aspx).
The highest incidence of gastric cancer can be found in southeast and central Asia, in regions of central Europe and in South America (Nagini, 2012). The occurrence of gastric cancer is between two and four times greater in men than in women, however, the course of the disease is similar in both sexes. Different factors have been associated to a greater risk of developing this pathology, among these is the high levels of salt intake, family history of gastric cancer, poor socioeconomic conditions, ancestry and others, however, the main factor associated with the development of gastric cancer is infection by H. pylori (Correa & Houghton, 2007; Dinis-Ribeiro et al. 2012). This association was discovered through serology studies of cases and controls, in which it was observed that the presence of H. pylori antibodies conferred a risk between 2.1 and 16.7 times greater of developing gastric cancer compared to seronegative individuals (IARC, 1994). H. pylori infects half of the global population with marked differences between regions (Suerbaum & Michetti, 2002). The prevalence of the infection in developed countries is between 20 and 40%, while in developing countries it is closer to 100% (Windsor et al. 2005). The bacteria causes gastritis in all the people infected, which may last decades unless the infection is treated, this gastritis can become a peptic ulcer in 10 to 20% of individuals and gastric cancer in 1 to 3% (Kuipers et al. 1995). The factors which determine that a subgroup of individuals infected with H. pylori develops a serious gastroduodenal disease while the majority remain carriers, are not completely clear. It is believed that it is a combination of environmental factors, the host´s genetics and the virulence of the bacteria.
The virulence locus of H. pylori most strongly associated with clinical disease compared to the asymptomatic disease is the cag pathogenicity island (cagPAI) (Yamaoka et al. 2008). The purpose of this revision is to describe the cagPAI of H. pylori, its effects on epithelial cells, variations in its genes and possible usefulness as preneoplastic lesion and gastric cancer markers.
MATERIALS AND METHODS
This is a narrative review, which was prepared by consulting the MEDLINE database between the years 1996 and 2016, using MESH describers: Helicobacter pylori, genomic islands, CagA protein and stomach cancer. Documents which addressed H. pylori generalities, clinical results of infection, epidemiology, pathogenicity island effects in intracellular signaling pathways and polymorphisms in pathogenicity island genes in their content were selected. The search was completed by reading and tracing bibliography referenced in the selected documents.
RESULTS
Helicobacter pylori. H. pylori is a gram-negative bacteria that was discovered as the main cause of chronic gastritis by Warren and Marshall in the year 1984, in a project which subsequently led them to become the winners of the Nobel Prize in Medicine, in 2005 (Marshall & Warren, 1984). It is a curved, multi flagellated, bacillary microorganism, which can also appear in coccoid form which is viable but not cultivable and is considered a dormant state (Kusters et al. 2006).
This bacteria is capable of surviving in the acid conditions of the stomach due to the action of the urease, an enzyme it has in abundance and which allows it to hydrolyze the gastric urea and produce ammonia (NH3) alkalinizing the surrounding medium (Miller & Maier, 2014; Smoot et al. 1990).
H. pylori and gastroduodenal disease. All the individuals infected with H. pylori develop superficial gastritis. In some individuals the gastritis is predominantly in the gastric antrum and is associated with a greater production of hydrochloric acid, it does not imply a greater risk of gastric cancer and is associated with a greater risk of duodenal cancer (Hansson et al. 1996). Another group of individuals develops gastritis that is predominantly in the gastric corpus which is associated with hypochlorhydria, gastric atrophy and a greater risk of gastric cancer (Kusters et al. 2006). Gastric cancer, specifically the intestinal type, is the result of a series of sequential steps described by pathologist Pelayo Correa, which begin with infection of the gastric mucosa by H. pylori and the transition from normal mucosa to chronic superficial gastritis which progresses to atrophic gastritis, intestinal metaplasia, dysplasia and finally gastric adenocarcinoma (Correa, 1996) (Figure 1).
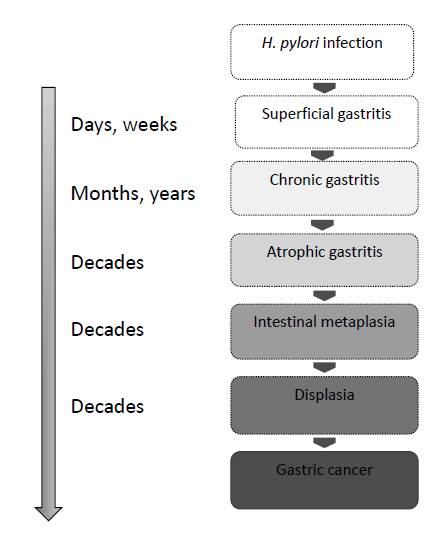
The result of the infection depends on environmental factors, the host and the bacteria. The virulence factor of H. pylori that is more strongly associated to clinical disease compared to asymptomatic disease, is the cagPAI (Hatakeyama, 2004).
cag Pathogenicity Island (cagPAI). Pathogenicity islands (PAI) are genomic elements, which carry one or more virulence genes, and are present in the genome of pathogenic bacteria, but absent in non-pathogenic isolates of the same species, generally occupying relatively big genomic regions, between 100 and 200 kb. The PAI have a percentage of guanine and cytosine different from the genome of the bacteria that carries them. It is considered that these PAI, which obtain the bacteria by horizontal transmission, maintain the basis composition of donor species. They are normally located near transfer RNA genes and are generally flanked by direct repeats (Schmidt & Hensel, 2004).
The pathogenicity island for H. pylori was described in 1996, its presence divides strains into two categories: very virulent or type I, which carry the cagPAI and mildly virulent or type II which do not carry the cagPAI (Censini et al. 1996). The cagPAI has an approximate size of 40 kb, it is flanked by 31 pb direct repeats, with a 35% content of guanine and cytosine unlike the bacteria which has 39% (Tomb et al. 1997), it contains between 27 and 31 genes. It was acquired horizontally by the bacteria, at the integration point of the cagPAI in the bacteria there are no transfer RNA genes, the cagPAI insertion in the bacteria interrupts the glutamate racemase gene (Akopyants et al. 1998; Censini et al. 1996). In some strains the cagPAI is divided into two segments I (14 genes) and II (16 genes) separated by an IS605 insertion sequence or in a few strains by a chromosomal sequence (Akopyants et al. 1998; Censini et al. 1996). In figure 2, there is a diagram of the cagPAI organization.
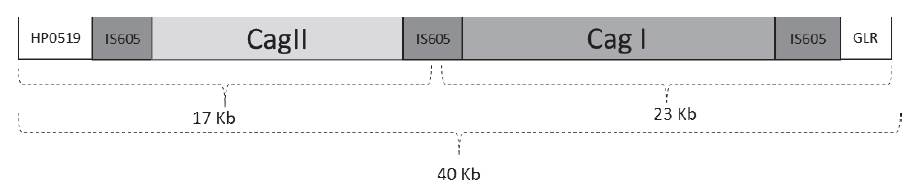
Figure 2 cag pathogenicity island structure flanked by glr and Hp0519 genes. The pathogenicity island is divided into two regions (cagII and cagI) separated by the presence of IS605 insertion elements. The genes contained code for the type IV secretion system.
The cagPAI genes code a type IV secretion system (T4SS), these systems are found in several bacteria, gram-negative as well as gram-positive, are evolutionarily related to DNA conjugation machineries and are used by pathogenic bacteria such as Agrobacterium tumefaciens, Bartonella, Bordetella, Helicobacter pylori and Legionella to introduce DNA or proteins into the eukaryotic cells. Consisting of approximately 11 virB proteins codified by virB1 to virB11 genes in the SST4 of the A. tumefaciens, considered the prototype (Backert et al. 2015; Fischer, 2011). The cagPAI shows significant differences to the T4SS of other bacteria, although it contains proteins that have similarities in sequence with all virB proteins, it has five important components that are not similar to any known protein (Backert et al. 2015; Fischer, 2011).
In figure 3, there is a diagram with the composition of the T4SS, at least 18 of the cagPAI genes code components of the H. pylori T4SS (Backert et al. 2015; Hagymasi & Tulassay, 2014). The components of this secretion system include: a) Pilus component: CagC, which forms the main extracellular structure that joins with the CagL, CagH, and CagI on the tip to interact with the β1 integrins; b) CagH, CagM, CagN, CagT, CagV, CagU, CagW, CagX, CagY, Cagδ central complex proteins that form the internal nucleus of the pilus; c) Cag/ and CagE energetic factors; that are ATPases that supply the system with energy (Tegtmeyer et al. 2011); d) Factors associated to translocation: GagF, CagZ, Cag/. e) Lytic Transglycosylases: Cag/. f) Substrate: CagA
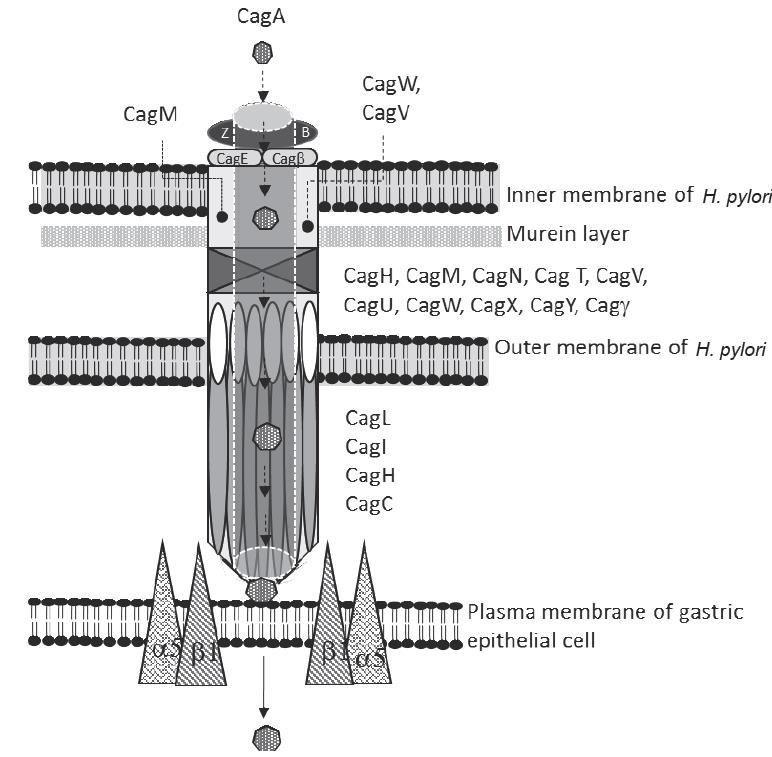
Figure 3 H. pylori type IV secretion system structure model. This is a complex formed by proteins encoded in the genes contained in the cagPAI of the bacteria that allow H. pylori to transfer CagA protein from the bacteria into the gastric epithelial cell.
Alteration of intracellular signaling pathways mediated by the cagPAI. The components of the H. pylori T4SS assemble to form a needlelike structure that emerges from the surface of the bacteria (Akopyants et al. 1998). Upon entering in contact with the epithelial cell, the CagL protein, an important component of the pilus, works as a bacterial adhesin that joins with the 5 1 integrins and activates it (Wiedemann et al. 2012). This event allows the transfer of the CagA protein from the bacteria, through the T4SS to the inside of the epithelial cell; where it interacts with about 24 cellular proteins dependently or non-dependently of phosphorylation (Backert et al. 2015; Mueller et al. 2012; Selbach et al. 2009; Tegtmeyer et al. 2011). This fact leads to the activation of signaling pathways which promote cellular responses with carcinogenic potential (Hatakeyama, 2004). Once the protein CagA comes into the cell is phosphorylated by Src and Abl kinases (Mueller et al. 2012). Phosphorylated CagA activates SHP2 phosphatase which in turn activates Erk 1/2 pathway and causes a reorganization of actin, cell dispersion and cell elongation, morphological changes similar to those caused by growth factors and which have been named hummingbird phenotype (Higashi et al. 2002a; Higashi et al. 2002b). The phosphorylated CagA protein can join with the Csk kinases and inhibit Src generating a negative circuit, which regulates the amount of phosphorylated CagA (Tsutsumi et al. 2003). In addition, the interaction of CagA with SHP2 inactivates the focal adhesion kinase FAK which can activate the Src (Tsutsumi et al. 2006).
Non-phosphorylated CagA protein also causes alterations in cell motility and proliferation through the binding to GRB/SOS/RAS and activation of the Raf/MEK/Erk pathway, it also joins with ZO-1 and JAM-A tight junction proteins and the E-cadherin protein of adherens junctions and causes deregulation of junction complexes (Amieva et al. 2003; Franco et al. 2005). The non-phosphorylated CagA interacts with c-Met and PLCy phospholipase activating proinflammatory and mitogenic responses (Churin et al. 2003), it also joins with the PAR1b cell polarity regulator and inhibits it which induces a loss of cell polarity (Saadat et al. 2007). It has recently been described that CagA triggers antiapoptotic responses through the interaction with the p53 stimulating protein and apoptosis (ASPP2) which activates p53 and induces apoptosis after DNA damage, CagA deregulates ASPP2 and causes the degradation of p53 through proteasome (Nesic et al. 2014). Another effect mediated by the joining of the T4SS to the cell is the induction of proinflammatory cytokines; once in the cell CagA can also induce a proinflammatory response which results in the expression of cytokines such as IL-8 through nuclear factor kappa B activation (NF-kB) (Brandt et al. 2005). In addition to CagA, H. pylori peptidoglycan can also be introduced in the cell through the T4SS, where it activates the NOD1 and the signaling pathways, which in turn activate NF-kB and production of proinflammatory cytokines such as IL-8, MIP2, and type 1 IFN (Viala et al. 2004).
Variations in the cagPAI Genes. The analysis of the sequence of the entire cagPAI for 29 strains from all the known biogeographical regions for H. pylori, reported by Olbermann et al. (2010), showed that the gene content as well as the organization of the cagPAI are well preserved. The diversity seen in cagPAI genes mostly consist in synonymous polymorphisms (Olbermann et al. 2010). However, some genes coding proteins that are secreted and located on the external part of the bacteria, have a high frequency of non-synonymous polymorphisms, among these are cag/, cagC, cagL y cag/. In cagC the codons with greater diversification are in amino acids 21 to 42, which are the ones exposed on the surface with highly specific strain epitopes in the N-terminal of the mature CagC protein. The cagY gene had variable lengths due to the presence of repetitive modules in the gene.
In a recent study of Rizzato et al. (2012), the cagC, pilus cagL, cagV, cagT and cag/ genes of the central complex and cagE that encodes one of the enzymes that supply the system with energy, were sequenced. These genes were chosen because their products are essential of the T4SS and some are exposed extracellularly (cagL and cagC). These genes were sequenced from the DNA obtained from 95 biopsies of Venezuelan and Mexican patients infected with H. pylori, diagnosed with gastritis (49 isolations) or gastric cancer (46 Isolations). In total 281 variants were found, observed in at least 10% of the samples originally analyzed. Then the frequencies of these genetic variants were compared between the cases of chronic gastritis vs. cases of gastric cancer. Twentysix single nucleotide polymorphisms (SNPs) located in the cagA, cagC, cagE, cagL, cagγ genes were found, 11 synonymous and 14 non-synonymous, which presented significant differences. Two polymorphisms in the cagE gene in positions 1039 and 1041 showed a highly significant association to cancer (p=2,07 x 10-6). Since it is possible that the strains from cancer represent simply those capable of surviving in the environment generated by the neoplasia and considering that the gastric cancer arises from a cascade of pre-malignant lesions induced by the infection with H. pylori: atrophic gastritis, intestinal metaplasia and dysplasia (Correa & Houghton, 2007); it is very important to evaluate these polymorphisms in samples from different pre-malignant states, and confirm their importance in our population.
cagY is one of the essential genes for T4SS functionality, measured through the ability to induce IL-8. CagY is a 220 Kd protein that is believed to facilitate contact between internal and external parts of the bacterial membrane (Kutter et al. 2008). The cagY gene has a structure that is not common, with multiple direct repeats of DNA located in a small region of 5’ repeats (IRR region) and in a large intermediate region of the gene (MRR region). Possible rearrangements of predicted DNA from these repeats invariably generate in-frame insertions or deletions which result in variant proteins. It has been recently demonstrated that the experimental infection with H. pylori in mice or monkeys induces recombinant events in cagY dependent on the host´s immunity, these changes induced in cagY can activate or inactivate the ability of the bacteria to induce IL-8, suggesting that the function of cagY diversity is to regulate the activity of the T4SS (Olbermann et al. 2010). The characterization of the cagY gene in samples from different progression stages of gastric cancer could show us if there are rearrangements in this gene associated with a greater possibility of the infection with H. pylori resulting in premalignant lesions and finally leading to gastric cancer.
CagA protein is phosphorylated in various Glu-Pro-Ile-TyrAla (EPIYA) sites present in its C-terminal region (Stein et al. 2002). Four types of EPIYA segments have been identified according to the sequences that flank the motifs (EPIYA-A, -B, -C and -D) (Higashi et al. 2002a). H. pylori isolates of the western hemisphere present the EPIYA-A and EPIYA-B segments followed by a variable number, from 1 to 3, of the EPIYA-C segment. In H. pylori from east Asia the CagA protein also presents the EPIYA-A and EPIYA-B segments followed by an EPIYA-D segment, that is only found in Asian isolations (Higashi et al. 2002a). However, in western as well as in east Asian strains additional variations in the distribution of the EPIYA segments have been found, in which the A or B segments can be duplicated or absent (ACC, ABCABC, ABDABD) (Naito et al. 2006; Reyes-Leon et al. 2007). In experiments in which CagA genes with different variants in EPIYA repeats are transferred to AGS epithelial cells, the SHP2 protein has been found to join with EPIYA C and D motifs in a phosphorylation dependent way (Higashi et al. 2002a). D motifs have greater affinity by SHP2 than C motifs; however, the greater the number of EPIYA C present in CagA the greater the efficiency in the binding with SHP2 (Higashi et al. 2002a). The ability of CagA to deregulate SHP2 seems to be important in determining the oncogenic potential of the strains. In some studies, a greater number of EPIYA-C segments have been observed in CagA protein of isolates from atrophic gastritis and gastric cancer cases compared to isolates from chronic gastritis, suggesting an association between the size of the CagA 3’ variable region and the clinical result of the infection (Argent et al. 2004; Azuma et al. 2002; Basso et al. 2008). However, in our studies and other studies, an association is not confirmed (Acosta et al. 2010; Reyes-Leon et al. 2007). On the other hand, it has been determined that the ability of CagA to interact with Csk is proportional to the number of EPIYA A and B segments, the capacity of CagA to join Csk attenuates the effects of its interaction with SHP-2 phosphatase and probably reduce its possible oncogenic effects (Naito et al. 2006).
DISCUSSION
H. pylori represents a very successful human pathogen with the potential to cause serious diseases in a subgroup of infected individuals. Research on cagPAI and CagA protein have led to the knowledge of mechanisms of pathogenesis of the bacteria. Several studies indicate that the CagA protein simulates a eukaryote-signaling factor located in multiprotein complexes or simultaneously in several areas of the infected cell and is capable of joining a number of factors, affecting several signal transduction networks that can have a significant impact on the sequence of the events that lead to gastric cancer.
In our country we have found that H. pylori infects over 77% of the adult population (Trujillo et al. 2014), despite this high prevalence of the infection, gastric cancer is present in a minimum percentage of the infected population. Therefore, it is important to identify the factors that determine that only a subgroup of infected individuals develops a serious gastroduodenal disease while the majority remain carriers.
In epidemiological studies it has been shown that the infection with H. pylori strains that carry the cagA gene is associated with a greater risk of developing peptic ulcer and gastric cancer (Yamaoka et al. 2008), however, in studies carried out by our group, we have found that 50% of individuals with superficial gastritis in our country are infected with strains that carry the cagA gene (Quiroga et al. 2005), Therefore, the mere presence of cagA gen or cagPAI does not allow us to predict the clinical result of the infection, mainly in high risk populations, such as the Colombian population.
Many of the functions of the CagA protein reside in a series of Glu-Pro-Ile-Tyr-Ala amino acid repeat motifs, called EPIYA motif, organized in tandem. Although it has been suggested that the presence of various copies of the type C EPIYA motif is associated with a risk of cancer and with greater activity of CagA in vitro (Higashi et al. 2002a), there is controversy regarding this (Rizzato et al. 2012). We have not found an association between the presence of more type C EPIYA motifs and the risk of gastric cancer in our analyses (Acosta et al. 2010; Fajardo et al. 2013).
Given the relevance the cagPAI has in the biological activities of H. pylori which lead to tissue damage, polymorphisms in the genes that code the proteins which form it, probably play a role as virulence factors. Twentysix polymorphisms have been described in the cagPAI cagA, cagC, cagE, cagL, cagT, cagV, and cagϒ genes, which present significant differences in their frequencies between gastric cancer and gastritis. It is important to evaluate these polymorphisms in preneoplastic lesions to examine if these variants also present significant differences in frequency between premalignant lesions and gastritis, so they could be used to identify infected individuals at risk of developing gastric cancer, who would be candidates for eradication.
In conclusion, the study of variations in cagPAI genes and their association with the development of preneoplastic lesions and gastric cancer could generate possible molecular markers of H. pylori virulence which would allow us to predict the risk of gastric cancer and preneoplastic lesions (atrophic gastritis, intestinal metaplasia, gastric dysplasia).

