











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCCIÓN
El cultivo de tomate (Solanum lycopersicum L.) es uno de los más importantes en el mundo, con una producción anual de 182 millones de toneladas (FAOSTAT, 2019). Para el 2018, en Colombia, se obtuvieron 183.419t, de las cuales, Antioquia aportó 5.327t y un rendimiento promedio de 28t/ha (Agronet, 2019). En la última década, los reportes de enfermedades de origen viral en tomate alrededor del mundo han aumentado, debido a diversos factores, tales como: la multiplicación vegetativa del cultivo, el movimiento internacional de semillas, plántulas y frutos, las siembras en nuevas áreas de cultivo, el calentamiento global que aumenta los ciclos de reproducción, la actividad de insectos vectores y el desarrollo de nuevas tecnologías de diagnóstico viral (Hily et al. 2018). Hasta el momento, se reportan en el mundo unas 136 especies de virus infectando el tomate (Muñoz-Baena et al. 2017a), un número muy alto comparado con otro tipo de solanáceas, como papa (Solanum tuberosum) (54 virus) y pimentón (Capsicum annuum) (49 virus) (Hanssen et al. 2010). En Colombia, se han reportado, en tomate, virus pertenecientes a los géneros: Begomovirus, Tobamovirus, Cucumovirus, Tospovirus, Crinivirus, Potyvirus y Nepovirus (Muñoz-Baena et al. 2016; 2017a).
El avance en las tecnologías de secuenciación de alto rendimiento (HTS, High-throughput sequencing) ha logrado impactar diferentes áreas del conocimiento; la sanidad vegetal fue una de las primeras beneficiadas, permitiendo la identificación de más de 100 nuevos virus de plantas (Adams et al. 2018). Utilizando esta tecnología de HTS, en Colombia, se ha logrado la identificación y la caracterización del genoma de un nuevo ilarvirus, infectando uchuva (Physalis peruviana), con el nombre propuesto: Cape gooseberry ilarvirus 1 (CGIV-1) (Gallo et al. 2018); la nueva variante de Potato virus S (PVS), a partir de papa criolla (Solanum phureja) (PVSP) (Vallejo et al. 2016); el primer reporte de Potato yellow vein virus (PYVV), Cucumber mosaic virus (CMV) y Alstroemeria necrotic streak virus (ANSV), infectando lulo (Solanum quitoense) (Gallo et al. 2018) y la secuenciación completa de dos variantes del Bell pepper endornavirus (BPEV), en pimentón (Muñoz-Baena et al. 2017 b). La HTS ha proporcionado una alternativa poderosa para la caracterización de genomas, sin conocimiento previo de las especies o variantes virales a evaluar, a diferencia de los métodos tradicionales de detección de virus, como RT-PCR, RT-qPCR o ELISA, que dependen de la disponibilidad de anticuerpos o cebadores (Maree et al. 2018).
Utilizando la secuenciación de alto rendimiento, a partir de ARN de doble cadena (ARNdc) de muestras de plantas de tomate var. Chonto, con síntomas típicos de enfermedades virales, en este estudio, se pretendió continuar con la identificación y la caracterización genómica de los virus que afectan el tomate en Antioquia, así como determinar su prevalencia, mediante pruebas de RT-PCR, en tiempo real (RT-qPCR) y RT-PCR, convencional. Adicionalmente, se evaluó la sanidad viral de la semilla sexual y de sus respectivas plantas derivadas, como base para establecer la importancia epidemiológica de este material en la transmisión de virus, en los cultivos de tomate de Colombia.
MATERIALES Y MÉTODOS
Material vegetal. Se obtuvieron muestras de tejido foliar de plantas de tomate var. Chonto, procedentes de cuatro lotes diferentes de Marinilla (Antioquia) (vereda El Rosario, N6°13’12.226”-O75°17’21.806”; vereda El Pozo, N6°13’11.028”-O75°15’ 43.022”; vereda El Porvenir, N6°14’7,513”-O75°16’27,286”; vereda La Peña, N6°13’19,141”-O75°18’2,216”), que presentaban síntomas de enfermedades virales (Figura 1), para la realización de HTS. Para la evaluación de prevalencia viral, se realizaron muestreos aleatorios en seis lotes de tomate, en los municipios de Marinilla y El Peñol. En cada lote, se obtuvieron tres muestras conformadas por cinco folíolos jóvenes para la posterior extracción de ácidos nucleicos. Con el fin de evaluar la prevalencia viral en semilla, se obtuvieron cinco muestras de semilla comercial (SC) de diferentes proveedores y cinco muestras de semillas no comerciales (SNC). Estas últimas semillas fueron extraídas de cuatro frutos maduros, obtenidos de diferentes mercados locales, luego de ser fermentadas durante tres días, secadas a temperatura ambiente y tratadas con jabón e hipoclorito al 1%, por 24h. De cada muestra de semillas, se hicieron tres réplicas y cada réplica fue dividida en dos partes: una, para la extracción de ARN total y, la otra, fue sembrada individualmente en bolsas con aproximadamente 500g de suelo y mantenidas bajo condiciones de casa-malla antiáfidos, por cinco semanas. Los brotes de las plántulas derivadas de cada tipo de semilla (brotes comerciales - BC y brotes no comerciales -BNC) fueron posteriormente utilizados, para la extracción de ARN total.

Figura 1 Síntomas asociados a enfermedades virales, observados en plantas de tomate var. Chonto, en el Oriente de Antioquia: a) Amarillamiento; b) Mosaico rugoso y deformación foliar; c) Bronceado; d) Mosaico rugoso con deformación en la base del pecíolo; e) Clorosis intervenal y, f) Enrollamiento foliar.
Secuenciación de alto rendimiento (HTS) y análisis bioinformáticos. Siguiendo el protocolo reportado por Valverde et al. (1990), se procedió a la extracción de ARNdc, a partir de 5g de tejido foliar sintomático, utilizando celulosa de fibra media (C6288 Medium) (Merck KGaA, Alemania), equilibrada en 16,5% de buffer STE (100mM NaCl, 10mM Tris-Cl, pH 8,0 y 1mM EDTA). Para la elución final, se emplearon 200µL de agua tratada con DEPC. La presencia de ARNdc fue verificada mediante la visualización de bandas, en un gel de poliacrilamida al 8%, teñido con GelRed (Biotium, EEUU). Posteriormente, se procedió a su desnaturalización a 98°C por 5 min y se evaluó su concentración y valor de RIN (RNA Integrity Number), en un equipo Bioanalyzer 2100 (Agilent Technologies, EEUU). La librería de ADN complementario (ADNc) fue construida con el kit TruSeq Stranded Total RNA LT Sample Prep, con eliminación del ARN ribosomal (ARNr), utilizando Ribo-Zero (Illumina, EEUU). La secuenciación fue realizada en un equipo Illumina NovaSeq de la compañía Macrogen (Corea del Sur). Una vez obtenidas las secuencias, se determinó su calidad con el programa FastQC y los nucleótidos de los extremos con baja calidad (QC<30) fueron eliminados, empleando el programa Trimmomatic v.0.36 (Bolger et al. 2014). Posteriormente, se realizó el alineamiento de los reads contra el genoma de referencia de tomate disponible en NCBI, utilizando el programa Bowtie2 (Langmead & Salzberg, 2013), con el fin de eliminar los reads de origen vegetal. De esta forma, los reads no alineados fueron ensamblados de novo, con el programa Trinity (Hass et al. 2013). Los contigs de origen viral, se identificaron por comparación con la base de datos de virus de referencia del NCBI, utilizando BlastN local. Los virus detectados, se verificaron y se ensamblaron por mapeo, utilizando Magic-Blast y los alineamientos resultantes, se visualizaron con el programa Tablet. Las secuencias consenso para cada virus fueron generadas con Samtools y los SNVs (Single nucleotide variants) fueron analizados con el programa LoFreq (Wilm et al. 2012). Finalmente, se identificaron los marcos abiertos de lectura (Open Reading frames - ORF), mediante BlastX y ORF finder. Para cada uno de los genomas virales identificados en este trabajo, se obtuvo la región codificante de la cápside viral (CP), para realizar análisis filogenéticos, utilizando el método de Máxima Verosimilitud, con el mejor modelo de sustitución, generado por el software MEGA7, con 1.000 réplicas de bootstrap (Kumar et al. 2016).
Prevalencia de virus por RT-qPCR. El ARN total de cada una de las muestras fue extraído con el kit GeneJET Plant RNA Purification Mini (Thermo-Fisher Scientific, EEUU), a partir de 100mg de tejido (foliar y semillas), macerado con nitrógeno líquido, siguiendo las instrucciones del fabricante. El ARN fue eluido en 40μL de agua tratada con dietilpirocarbonato (DEPC). La concentración e integridad del ARN purificado fue evaluada por lecturas de absorbancia, a 260 y 280nm, en un equipo Nanodrop 2000C (Thermo-Fisher Scientific, EEUU). A partir de este ARN, se procedió a la síntesis del ADNc para los virus: PVY, PVS, PYVV, PVX y STV, utilizando 100pmol de cada cebador reverso (Tabla 1). Para PVY, PVS y PVX, se empleó el cebador Oligo-(dT)18; para PYVV, el PYVV_R_CP y para STV, el STV-R. Las reacciones de ADNc, se hicieron en 20μL, conteniendo 200U de la enzima RevertAid Reverse Transcriptase (Thermo-Fisher Scientific, EEUU), 1X de buffer RT, 0,5mM de los cuatro dNTPs, 20 U de inhibidor de ARNasas RiboLock (Thermo-Fisher Scientific, EEUU) y de 100 a 500ng de ARN. Las reacciones fueron incubadas a 42°C durante 60min y, posteriormente, la enzima se inactivó a 70°C, durante 10min. Para la qPCR, se utilizó el kit Máxima SYBR Green/ROX (Thermo-Fisher Scientific, EEUU), en un volumen total de 12,5μL, con cebadores específicos, a una concentración de 0,3μM (Tabla 1). El programa de amplificación consistió de una activación de la Taq polimerasa, por 10min, a 95°C, seguido de 35 ciclos, a 95°C, por 15s y 50°C, por 45s, utilizando un equipo Rotor-Gene Q-5plex (Qiagen, Alemania), para calcular los valores de ciclo umbral (Ct) y de temperatura de fusión (Tm), en el rango 50-99°C. Todas las reacciones tuvieron un control positivo y un control negativo, que correspondía a tejido foliar infectado (+) o libre (-), de cada virus bajo estudio.
Tabla 1 Secuencias de los cebadores utilizados en el estudio para la síntesis del ADNc, RT-qPCR y RT-PCR.
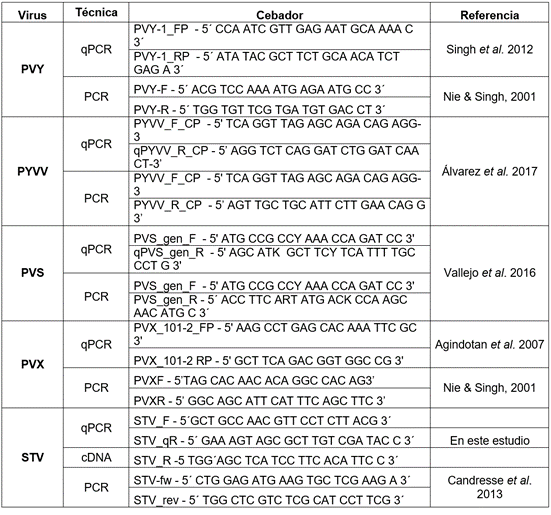
Identificación por RT-PCR convencional y secuenciación Sanger. Las reacciones de RT-PCR, se realizaron para amplificar la cápside de cada virus, con cebadores específicos (Tabla 1), en un volumen de 25µL, siguiendo las condiciones descritas por Gallo et al. (2018). Al menos tres bandas de muestras individuales del tamaño esperado para cada virus fueron purificadas, con el kit GeneJET Gel Extraction (Thermo-Fisher Scientific, EEUU) y secuenciadas en ambos sentidos, por el método de Sanger, en un equipo ABI Prism 3730xl, de la compañía Macrogen (Corea del Sur), para su posterior análisis filogenético.
Análisis estadístico. Para el análisis estadístico, se utilizó, como variable respuesta, los valores de ciclo umbral (Ct) de las muestras que resultaron positivas, según su temperatura de desnaturalización (Tm). Para el caso de las evaluaciones de virus en semilla sexual, se realizó un ANOVA, con un nivel de significancia de p<0,05, utilizando la prueba Friedman, con el fin de detectar diferencias significativas para la detección de cada virus entre semilla y en sus brotes, derivados bajo condiciones de casa-malla y la prueba Kruskal-Wallis, para las comparaciones entre muestras de semilla comercial (SC) y no comercial (SNC). Estos análisis, se realizaron con el programa RStudio (RStudio Team, 2015).
RESULTADOS Y DISCUSIÓN
Secuenciación de alto rendimiento (HTS). Del análisis HTS del ARNdc de tejido foliar sintomático de tomate, se obtuvieron 15.916.085 reads pareados, de los cuales, alrededor del 39%, correspondieron a los ARN derivados del genoma vegetal, mientras que del 61% de los restantes (9.732.192), 786.824 reads fueron de origen viral y correspondieron a genomas de crinivirus, amalgavirus, alphaendornavirus, potexvirus, potyvirus y carlavirus, siendo PVY, PVX, PVS, PYVV, BPEV y STV, las especies virales que presentaron mayores niveles de abundancia. PVY, PVS, PVX son virus flexuosos con genoma monopartita de ARN de cadena sencilla positiva (ARNcs+), frecuentemente, detectados infectando plantas de la familia Solanaceae. La transmisión de estos virus es mecánica, por propagación vegetativa y para el caso de PVY y PVS, por diversas especies de áfidos, usualmente, de forma no persistente y, en algunos casos, por semilla sexual (Cox & Jones, 2010; Gutiérrez et al. 2013; 2015; Hull, 2014).
El virus PYVV presenta tres segmentos genómicos individualmente encapsidados en partículas flexuosas; es transmitido de manera semipersistente, por la especie de mosca blanca Trialeurodes vaporariorum (Hemiptera: Aleyrodidae), siendo diferentes plantas de la familia Solanaceae sus principales hospedantes (Guzmán-Barney et al. 2013; Gallo et al. 2018). Finalmente, BPEV y STV son virus con genoma de ARNdc que, aparentemente, solo se pueden transmitir, a través de semillas sexuales. BPEV, se ha encontrado con frecuencia en arroz (Oryza sativa), cebada (Hordeum vulgare), frijol común (Phaseolus vulgaris) y pimentón (Capsicum annuum) (Muñoz-Baena et al. 2017b), mientras que STV, se ha reportado en tomate y, recientemente, en uchuva (Gallo et al. 2020).
En el set de datos, también se identificaron segmentos de genomas de micovirus (933 reads), asociados a los géneros Mitovirus, Botybirnavirus, Mycoflexivirus y a un micovirus de ARNdc, de género no asignado (2.218 reads); así como 100 reads provenientes del genoma de ADN de doble cadena, de un cavemovirus (Figura 2). Una vez realizado el ensamblaje de las secuencias virales, se obtuvieron los genomas completos de PYVV (ARN1: 7.829 nt, MN088360; ARN2: 5.296 nt, MN088361; ARN3: 3.815 nt, MN088362) y los genomas parciales de los virus PVY (9.663 nt) y PVX (6.423 nt). Al comparar los ensamblajes obtenidos con las secuencias depositadas en las bases de datos moleculares, se encontraron altos niveles de identidad, respecto a aislamientos colombianos de estos virus. Para PYVV, se encontraron niveles de identidad del 99%, con un aislamiento reportado en lulo (MG696858) (Gallo et al. 2018) y en papa criolla (KR998194) (Álvarez et al. 2017). Para el caso de PVY y PVX, se realizaron las comparaciones con sus genomas parciales, hallando 100% de identidad con aislamientos de la raza PVYNTN, obtenidas a partir de tubérculo-semilla de papa (MK041096, MK024932) y PVX presentó un 98% de identidad, con el genoma completo de un aislamiento de este virus en tomate, en Antioquia, ubicado en el linaje Eurasiático (MH282866). Asimismo, el análisis de HTS permitió el ensamblaje completo del genoma del STV (3.437 nt) y del BPEV (14.706 nt), lo que representa el primer reporte de estos virus en Colombia, para el cultivo del tomate. Para el caso del genoma de STV (MN095716), se identificaron dos ORF: el ORF 1, próximo al extremo 5´, que codifica para la proteína de cápside (CP) y el ORF 2, cercano al extremo 3´, con un cambio de marco de lectura +1, que codifica la RdRp; dicha secuencia compartió un porcentaje de identidad del 99%, con aislamientos procedentes de México (EF442780) y Estado Unidos (KX949574), lugares donde fue reportado, por primera vez, este virus (Sabanadzovic et al. 2009); aunque desde entonces se ha informado de su detección en otros países, como Francia, España, Canadá y China (Alcalá et al. 2017). De STV, solo se conoce su trasmisión, a través de semilla (Xu et al. 2017); aunque la presencia de este virus, generalmente, se asocia con plantas asintomáticas, se ha reportado que induce síntomas, como decoloración en los frutos y reducción del tamaño de las plantas, en algunas variedades de tomate (Puchades et al. 2017), por lo que la determinación de su efecto sobre la variedad Chonto, cultivada en Colombia, resulta prioritario, para evaluar su nivel de riesgo fitosanitario.
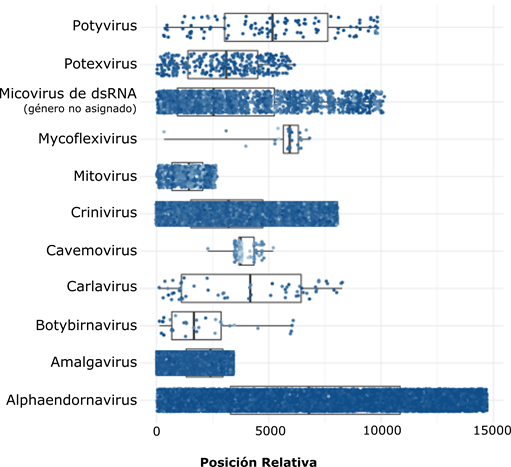
Figura 2 Identificación de virus de ARN, mediante HTS, a partir de ARNdc, procedente de tejido foliar de plantas de tomate con síntomas de infecciones virales, en Marinilla (Antioquia). Los puntos representan los reads alineados con los genomas de referencia; su densidad corresponde a la profundidad de la secuenciación y la intensidad del color indica el porcentaje de identidad de cada read, respecto a la referencia.
Por otro lado, el ensamblaje del genoma de BPEV (MN073197) permitió identificar su único ORF, que codifica para una poliproteína, que contiene los motivos metiltransferasa (MET), helicasa (HEL), glicosiltransferasa (GT) y RdRp. El genoma de este virus compartió un 99% de identidad con cepas de BPEV (NC_039216, KX977568, MK284997), obtenidas en pimentón, en Antioquia (Muñoz-Baena et al. 2017b; Gallo et al. 2019). Aunque la transmisión horizontal de los endornavirus no ha sido reportada, estos virus, a menudo, se encuentran en una gran variedad de plantas y con un alto porcentaje de transmisión vertical (semilla), manteniendo una relación simbiótica con su hospedante (Fukuhara, 2019).
Prevalencia de virus por RT-qPCR. La presencia de los virus identificados mediante HTS fue confirmada, empleando la técnica RT-qPCR, con cebadores específicos, para los virus PVS, PVX, PVY, PYVV y STV (Tabla 1). El virus BPEV no se incluyó en esta evaluación, debido a su característica simbiótica en las plantas (Fukuhara, 2019). Los resultados obtenidos por RT-qPCR sobre las 18 muestras de tejido foliar (3 plantas/6 lotes), mostraron una prevalencia del 100%, para PYVV, seguido de PVX (94,4%), PVY (77,8%), STV (72,2%) y PVS (5,6%) (Figura 3 a). Los valores de Ct indicaron baja variación en los niveles de título viral de las muestras evaluadas; el menor valor encontrado de Ct fue para PYVV (Ct=22,8; SD=5) y así, sucesivamente, para PVX (Ct=23,6; SD=2,7), STV (Ct=26,9; SD=4,2), PVY (Ct=28,7; SD=9) y PVS (Ct=35; SD=0), que solo fue detectado en los últimos ciclos de la reacción, en uno de los lotes evaluados. La comparación de estos resultados respecto a los previamente reportados para el oriente Antioqueño, en el mismo hospedante, evidencian el aumento aparente de la prevalencia de estos virus, pues el PVY, se había detectado en niveles del 13,3% (Muñoz-Baena et al. 2016) y el PYVV, en el 72% de las muestras analizadas, en dichos estudios (Muñoz-Baena et al. 2017a). Esta situación debe llamar la atención de los gremios de productores y organismos de sanidad vegetal del país, de manera que se emprendan programas de manejo integrado de enfermedades virales, que conduzcan a la reducción de estos altos niveles de infección en los cultivos de tomate, de esta región del país.
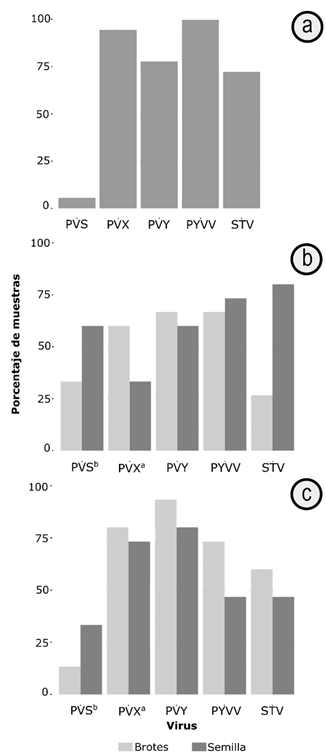
Figura 3 Porcentaje de muestras de tomate identificadas con virus: a) Muestras de tejido foliar de cultivos de tomate var. Chonto de Marinilla y El Peñol y b) Muestras de semilla comercial (SC) y no comercial (SNC) y sus respectivos brotes, derivados bajo condiciones de casa-malla. a y b indican diferencia estadística significativa (p<0,05), para la detección de PVX y PVS.
Para el caso de las evaluaciones de semillas (SC y SNC) y sus respectivos brotes (BC y BNC), se detectó la presencia de todos los virus bajo estudio, con valores de Ct, muy cercanos a los detectados en las muestras de campo (Ct=25,2; SD=3,1 a Ct=32; SD=3,2). En el caso de SC, se destaca la detección de STV, como el virus de mayor prevalencia (80%) (Figura 3 b), lo que no resulta inesperado, dado que se conoce de su transmisión vertical, a través de semilla de tomate, en niveles de hasta el 90% (Alcalá et al. 2017). El segundo virus con mayor nivel de detección en el material SC correspondió al PYVV, que se detectó en el 73% de las muestras evaluadas, un valor muy alto para un virus que, hasta hace unos años, se consideraba solo de importancia en cultivos de papa de la región andina de Suramérica, donde se ha reportado que puede causar pérdidas hasta del 50% (Franco-Lara et al. 2013). Dados los resultados encontrados en esta investigación será importante, en el futuro próximo, evaluar los efectos de este virus sobre la producción y la calidad de los frutos de tomate de la variedad Chonto, en nuestro país. La evaluación de los brotes derivados (BC) de SC indicó la presencia de los virus PYVV y PVY, como los de mayor prevalencia en dichos materiales, con un valor del 66%; mientras que para SNC y sus plántulas resultantes (BNC), se encontró la infección de los virus PVY y PVX, como los de mayor prevalencia, con niveles del 80 y 73%, en SNC y del 93 y 80%, en BNC, respectivamente (Figura 3 b). La veracidad de las pruebas de RT-qPCR fue confirmada por análisis de las curvas de desnaturalización, utilizando la herramienta HRM (High Resolution Melting). Los valores de Tm para todos los virus y para los diferentes tipos de muestras, se observaron en los rangos previamente reportados en Colombia, para PYVV (Tm=77,28 ±0,6°C) (Álvarez et al. 2017); PVX (Tm=80,3 ± 0,5°C y 83,7 ± 0,5°C) (García Ruíz et al. 2016); PVY (Tm=77,5 ±0,5°C) (Medina et al. 2015); PVS (Tm=86,96±1°C) (Vallejo et al. 2016) y, finalmente, para STV, el valor promedio de Tm fue de 82,3± 0,2°C. Ya que este es el primer registro de este virus en Colombia, se confirmó la naturaleza de sus amplicones, por secuenciación Sanger. Todos los controles positivos amplificaron la secuencia esperada con valores de Ct inferiores a 25 y no se observó amplificación en ninguno de los controles negativos.
Al comparar los resultados obtenidos entre el material de siembra de origen comercial y no comercial, se halló una diferencia estadística significativa para la detección de PVX (p=0,02) y PVS (p=0,04), encontrándose una mayor prevalencia de PVS, en el material de siembra comercial y de PVX, en el material de siembra de origen no comercial, lo que, posiblemente, está relacionado con las diferencias en la condición fitosanitaria de sus plantas madre. La comparación entre la detección de los virus bajo estudio en semilla y sus respectivos brotes, no mostró ninguna diferencia estadística, lo que indica que, para el establecimiento de un programa de certificación de semilla sexual de tomate, por su sanidad viral, la evaluación directa con RT-qPCR de estos virus sobre la semilla es suficiente y no se requiere la siembra para evaluación de plántulas. Esto representará un ahorro importante en el tiempo y en la logística requerida para la ejecución de dichos programas.
Por otra parte, llama la atención la prevalencia de infecciones mixtas entre los diferentes tipos de material de siembra de tomate (SC y SNC) (Figura 4), siendo detectados los cinco virus evaluados en el 7% de las muestras, tanto en los BC como en los BNC. Para el caso de SC, la combinación de mayor prevalencia fue PVY+PYVV+PVS+STV (33%), mientras que en BC, se encontraron infecciones mixtas de PVY+PVX+PVS y PVY+PYVV+PVX, en el 13% de las muestras. En los materiales de SNC, la mayor prevalencia encontrada correspondió a la combinación PVY+PYVV+PVX (27%), seguida por PVY+PVX (20%), mientras que en sus plantas derivadas BNC, la infección mixta más frecuente fue aquella de PVY+PYVV+PVX (27%).
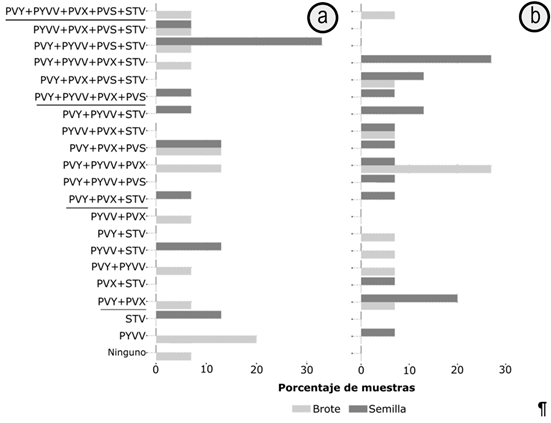
Figura 4 Infecciones mixtas en lotes semilla de tomate comercial (SC) y no comercial (SNC), en Antioquia y sobre los brotes de sus plantas derivadas.
En los últimos años, la implementación de las nuevas tecnologías de secuenciación masiva ha permitido ampliar el conocimiento de las infecciones mixtas en tejidos vegetales; es así, como recientemente, mediante HTS de ARN pequeños (sRNA), se logró la detección de los virus PVY, PVX, STV y del Pepino mosaic virus (PepMV), en diferentes solanáceas, incluyendo tomate (Turco et al. 2018). De igual manera, en China, utilizando HTS, se detectaron 22 especies diferentes de virus en cultivos de tomate, siendo muy relevante el hecho que el 89% de las muestras evaluadas presentaron la infección de dos o más virus (Xu et al. 2017). Sin duda, la alta frecuencia de infecciones mixtas detectadas en este trabajo, en material de siembra y en plantas en producción de tomate, señalan la necesidad de emprender evaluaciones que determinen si las interacciones entre los virus encontrados corresponden a sinergismos, antagonismos o relaciones neutrales, así como estimar los efectos de dichas combinaciones de virus sobre los rendimientos del cultivo de tomate en Colombia y en otros países.
Identificación de virus mediante RT-PCR/Sanger. A partir de los extractos de ARN, se obtuvieron amplificaciones de la región CP, para PVX (562 pb), PVY (480 pb), PVS (421 pb), PYVV (495 pb) y STV (864 pb). Los análisis filogenéticos de dichas secuencias, así como de las secuencias de CP obtenidas de los ensamblajes de HTS, ubicaron a PVX en el linaje Eurasiático, en conjunto con aislamientos previamente obtenidos en tomate y papa de Antioquia, así como también con secuencias provenientes de China e India (Figura 5). El análisis para PVY ubicó los aislamientos, aquí obtenidos, en los clados que representan la raza PVYNTN y PVYN de este virus, con niveles de identidad superiores al 99%, respecto a cepas obtenidas en papa, tomate de árbol, uchuva y tomate en Colombia. En el caso de PVS, las secuencias se ubicaron en el clado correspondiente al linaje Andino (PVSA) de este virus, junto con secuencias provenientes Antioquia y cuyo genoma fue obtenido en papa criolla en Antioquia (Gutiérrez et al. 2012). Para el virus PYVV, se presentaron niveles de identidad superiores al 99% entre las secuencias obtenidas y aquellas previamente reportadas en lulo y papa en Colombia. Finalmente, los aislamientos de STV presentaron niveles de identidad del 99%, respecto a cepas de este virus, procedente de tomate en diferentes continentes (Figura 5).
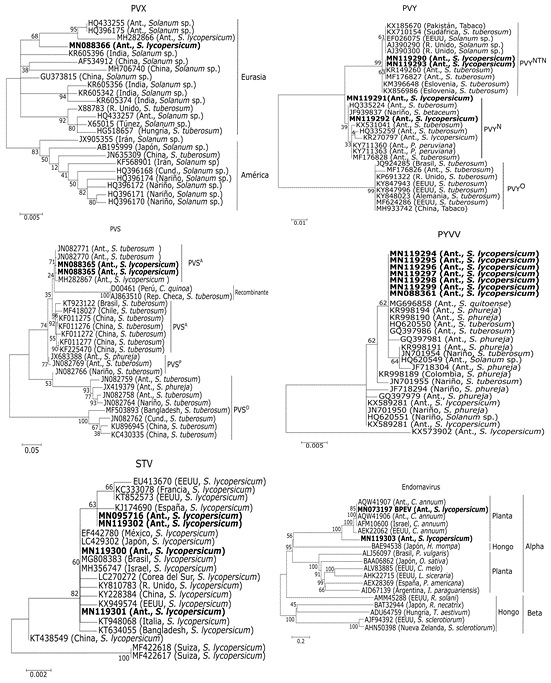
Figura 5 Árboles filogenéticos, obtenidos con las secuencias correspondientes a la región de la cápside, para los virus PVX, PVY, PVS y PYVV. Para STV, se empleó la secuencia codificante de RdRp y para el caso de los endornavirus, se realizó dicho análisis con la poliproteína. Los números sobre las ramas indican los valores de bootstrap y los nombres en la derecha representan las variantes o linajes reportados en la literatura, para cada especie viral.
Estos hallazgos, indican la posible ocurrencia de patogenicidad cruzada entre los virus de ARN evaluados en el Oriente de Antioquia y, por tanto, la necesidad de revisar los ciclos de rotación de cultivos, los arreglos de siembra mixtos y el establecimiento de cultivos escalonados de plantas solanáceas, tan frecuentes en esta región y en otras zonas andinas de Colombia, pues desde el punto de vista fitosanitario, dichas situaciones favorecen el aumento epidemiológico de las infecciones virales.

